Chapter 1 약동학 기초 이론
임동석
1.1 서론
1.1.1 용어의 소개
약동학(藥動學, pharmacokinetics, PK)이란 용어는 그리스어로 약을 의미하는 pharmacon과 움직임이라는 kineticos를 합하여 만든 것이다. 약동학은 약이 몸 안으로 흡수되고, 분포되었다가 대사, 배설을 통해 몸 밖으로 나갈 때까지 혈액과 각 조직에서 발견되는 약의 농도가 시간에 따라 쉼없이 변화하는(즉, 움직이는) 과정을 설명하는 방법이다. 용어만 들으면 마치 약물 분자들이 스스로 몸 속에서 움직여서 그 농도가 변하는 것으로 오해할 수도 있다. 그러나 실제로는 생체 장기와 조직들이 몸 밖에서 들어온 `약’이라 불리는 이물질을 다루면서 일어나는 작용들, 즉 장관에서의 흡수, 심장박동으로 일어나는 혈류에 의한 말초조직으로의 분포, 간이나 콩팥을 통한 몸 밖으로 제거 등이 혈액과 각 조직들에서의 약물의 농도가 올라갔다가 다시 내려가는 현상을 일으키는 원인인 것이다.
반면 약이 수용체에 결합한 후 일어나는 세포나 몸에서 일어나는 변화(약효)의 크기와 양상을 약물농도와의 관계로 설명하는 학문은 약력학(pharmacodynamics, PD: pharmacon에 힘을 의미하는 그리스어 dynamicos를 붙임)라 부른다. 이 두 방법론은 사람에서의 약효를 예측하고 적절한 용량용법(dosage regimen)을 찾기 위한 필수적인 도구로 쓰이고 있다. 약동학과 약력학을 구분하기 위해 흔히 쓰이는 정확하고 간결한 설명으로는 “몸이 약에게 하는 작용을 다루는 것이 약동학, 약이 몸에게 하는 작용을 다루는 것이 약력학” 이란 말이 있다. 그러나 이들 분야를 처음 접하는 사람이 들으면 무슨 뜻인지 이해하기 어려운 표현이다. 과거 약력학에 대한 연구나 이해가 적던 시절, pharmacokinetics를 약물동력학 이라고 번역하기도 했으나, 이 용어는 약력학과의 구분이 어려우므로 피하는 것이 좋다. 일본에서는 약물동태학이라고도 번역하고 있으나 약동학과 같은 의미이다.
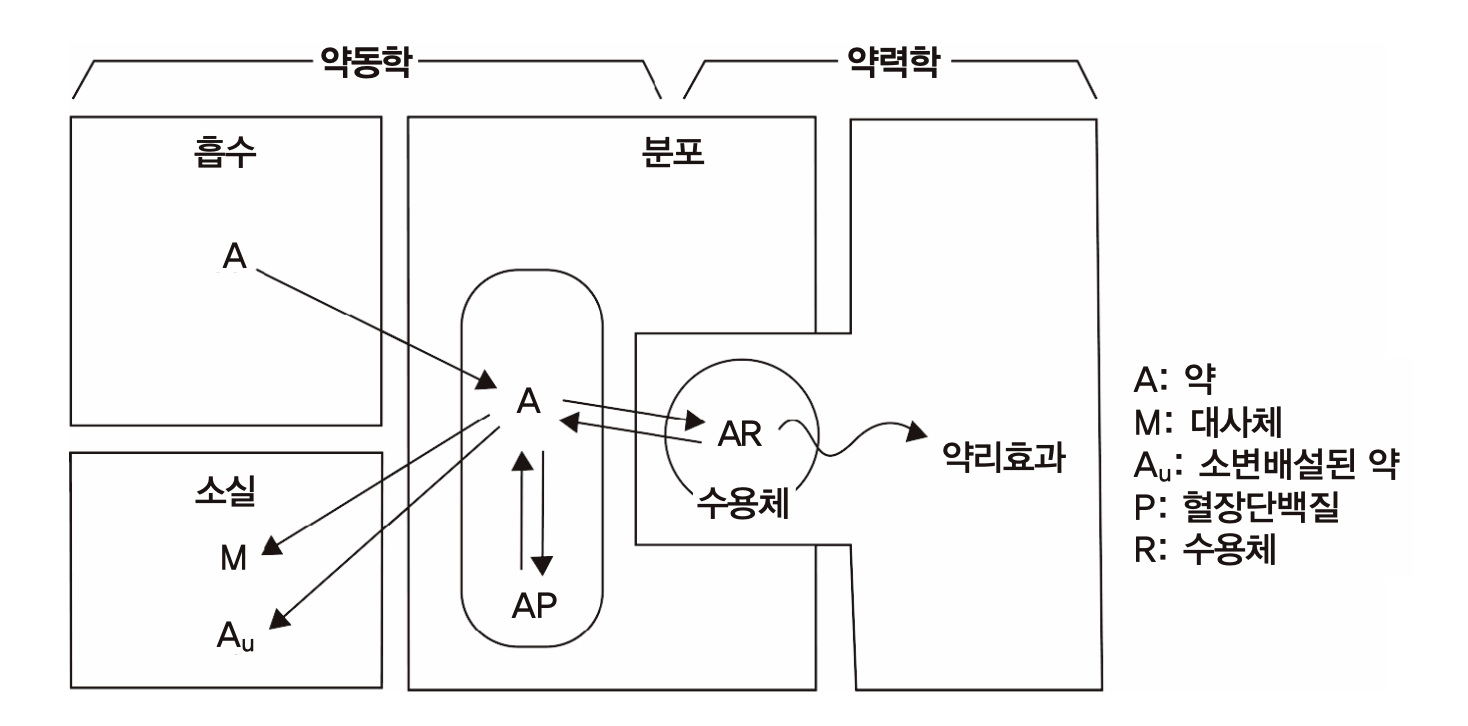
그림 1.1: 약동학과 약력학의 구분
1.1.2 약동학을 공부하는 목적
신약 한 가지를 발굴(discovery)에서부터 시판허가에 이르기까지의 과정에서 단계별로 수없이 많은 의사결정들을 하게 되는데, 이들 하나하나에서 약동학이 관련되지 않는 것이 드물다. 왜 약동학이 그토록 많이 적용되는가에 대한 답이 그림 1.2에 나와 있다. 약이 단순한 화합물이 아니라 약인 이유는 환자에게 주었을 때 효과가 있기 때문이다. 약 개발 과정에서의 의사결정을 단순하게 말한다면 약효를 적절히 나타내기 위한 용량을 찾아가는 과정이라 할 수 있다. 매 개발의 단계마다 약효나 그와 관련된 생체표지자 등의 변화를 측정하여 적절한 효과를 나타내는 용량을 선택하여 다음 단계의 연구를 수행하면 된다. 그러나 실제 사람에서 약효라고 부를 수 있는 것을 한 번의 투여 간격 사이에 여러 번 측정하는 것은 혈압이나 혈당과 같은 예외적인 생체표지자들을 제외하면 거의 불가능하다. 그러면 어떻게 적절한 용량과 그 투여간격을 정하는가? 이에 대한 답은 그림 1.2에 나타나 있다. 그림의 아래 패널과 같이 우리가 알고 싶은 것은 어떤 용량용법에서 약효가 어떤 범위 내에서 유지되는지에 관한 것이다. 그 범위를 적절하게 유지시켜주는 용량용법을 선택해야 하는데, 사람에서 약효를 쉽게 측정할 수 없기 때문에 대신 쓰는 방법은 그림 1.2의 윗부분과 같이 약동학과 약력학의 관련성을 이용하는 것이다. 그림의 위 우측 패널을 보면 in vitro 연구나 동물실험자료에서 얻은 약물농도와 생체표지자나 약효의 관계(약력학)가 나타나 있으므로, 얼마의 농도를 얻으면 얼마나 효과가 나타날지를 추정할 수 있다. 또 위의 왼쪽 패널에는 사람에서의 혈장 약물농도의 시간에 따른 변화 추이(약동학)가 나타나 있는데, 이는 임상시험에서 채혈을 통해 비교적 쉽게 확보할 수 있고, 비임상개발 단계에서는 각종 기법으로 예측할 수도 있다. 즉 윗부분의 두 패널의 정보를 결합하면, 사람에서 약을 먹은 후 몇시간 뒤에 약효가 어느 정도나 될지를 아래의 패널과 같이 예상할 수 있고, 결국 적절한 효과를 확보하기 위한 용량용법을 찾을 수 있다는 것이다. 다시 말하면, 실제 효과를 사람에서 관찰하기 어려우니 대신 가지고 있는 약물농도-약효의 관련성 정보(약력학)에다, 사람에서 비교적 쉽게 측정 또는 예측할 수 있는 약동학 정보를 결합하여 용량용법을 예측하는 것이며, 개발이 진행되면서 약동학, 약력학 정보가 추가됨에 따라 그 관련성에 관한 모델은 업데이트되지만, 약동-약력학을 결합하여 예측한다는 방법론 자체는 변치 않는다.
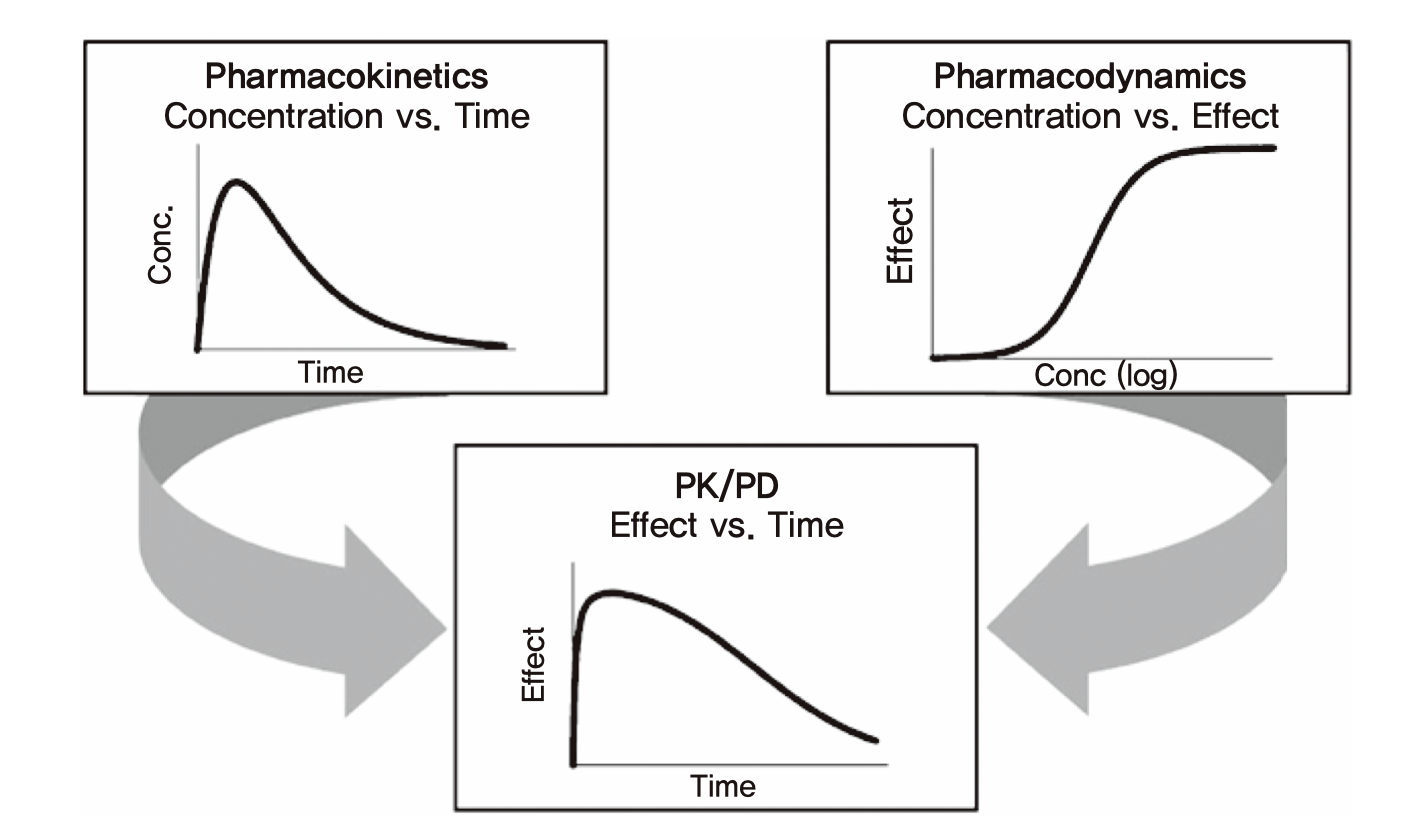
그림 1.2: 약동학과 약력학을 이용한 적절한 용량용법의 추정 원리 (Derendorf and Meibohm 1999)
그러므로 우리가 생리학과 약리학적 지식에 더하여 각종 수식을 써 가면서 만든 약동학 이론으로 사람에서의 혈장 약물 농도의 변화를 설명하고 예측하고자 하는 것은 사실, 용량과 약효의 연관성을 설명하고 예측하기 위함이다. 즉, 어떤 약의 약동학을 잘 안다 해도 그것만으로는 쓸모가 없고, 그 약의 혈장농도와 약효의 연관성에 관한 정보(약력학)가 확보되어 있을 경우에만 그 두 가지를 결합하여 개발과 인허가의 의사결정에 적극 활용할 수 있게 된다는 것이다.
1.2 ADME와 약동학
약동학을 아주 좁은 의미로 표현한다면 혈액 또는 혈장의 약물농도가 시간에 따라 변화하는 양상을 설명하는 모델을 찾는 것이다. 가장 일반적인 약동학 모델은 경구투여 후 선형적으로 체내에 흡수된 후 혈관 밖의 조직들로 분포됨과 동시에 선형적으로 몸 밖으로 제거되는 것이다. 그 과정들을 나타내는 약동학 파라미터들은 흡수에는 일차 흡수속도상수(ka)와 생체이용률(F), 분포에는 분포용적(Vd), 대사나 배설을 통한 제거에는 청소율(CL)이다. 즉 혈장약물농도는 몸 밖에서 투여한 약의 용량과 채혈시간 및 약동학 파라미터들인 ka, F, Vd, CL 에 의해 결정되는 종속변수인 것이다: C(t) = f(Dose, time, ka, F, Vd, CL) 괄호 안의 독립변수들 중 용량과 채혈시간은 외부에서 인위적으로 정할 수 있는 것이므로 약동학 파라미터라고 하지는 않는다. 이 절에서 각 PK 파라미터들의 의미와 흡수, 분포, 대사, 배설의 관련성을 간략히 살펴보기로 한다.
1.2.1 흡수(absorption)
흡수와 관련된 약동학 파라미터들은 ka와 F이다. 약을 물과 함께 삼키면 위장 속에 들어와서 소장으로 내려가면서 흡수의 과정을 거친다. 흡수가 일어나는 첫 과정은 알약 또는 캡슐 형태로 들어온 약이 소화액 속에서 부스러지고 용해된 후, 그 속에 녹아있는 약물 분자들이 장 상피세포들을 통과하는 것이다. 소화관 속에서 일어나는 이 붕해, 용해의 과정이 얼마나 빨리 일어나는지와 관계된 것이 일차흡수속도상수인 ka이다. 빨리 녹고 빨리 장 상피세포를 투과하여 핏속으로 들어가는 약일수록 ka가 크다. 약의 소화액에서의 용해도(solubility)와 장 상피세포를 투과하는 속도(permeability)는 반비례하는 경향이 있다. 예컨대 지용성이 좋은 물질일수록 물에 잘 안 녹으므로 알약 형태로 투여할 경우 소화액에 제대로 용해되기 힘들 것이다. 반면에 수용성이 좋은(극성이 강한)물질은 소화액에 잘 녹지만, 막상 인지질로 구성된 장 상피세포막을 통과하는 것은 극성으로 인해 매우 느릴 수 밖에 없다. 그러므로 수용성과 지용성의 어느 극단에 치우치지 않고 적당한 범위의 물성을 가진 물질이라야 먹는 약으로 개발할 가치가 있다. Lipinsky의 Rule of 5는 이런 물성을 요약해 놓은 것이다. 경구흡수가 떨어지는 물질들은 공통적으로 아래와 같은 물성을 가지는 경향이 있다는 것이다.
- more than five hydrogen bond donors (the sum of OHs and NHs)
- molecular weight greater than 500
- the logP over 5
- more than 10 hydrogen bond acceptors (the sum of Ns and Os)
실제 약물이 흡수되는 속도가 위장관 내에 남아있는 약물의 양에 비례하는, 1차식(first-order kinetics)을 따르는 경우는 드물고 소장을 따라 내려가면서 각 구역마다 소화액과 장 상피세포, 혈액 등에서의 약물의 농도, 이온화 정도 등의 차이에 의해 매우 복잡한 양상을 보이지만, 이를 일일이 반영하지 않고 최고혈중농도 예측 등에 어느정도 오류가 있음에도 불구하고 단순한 일차흡수모델을 가장 흔히 쓰고 있다.
약이 소장에서 장 상피세포를 통과하고 간문맥을 통해 간으로 들어갈 때까지는 아직 전신으로 흡수가 되었다고 할 수 없고, 간을 통과하여 심장을 통해 전신을 도는 순환혈액 속으로 들어갔을 때 비로소 흡수가 되었다고 한다. 이렇게 전신순환혈로 들어오기까지 세 가지의 관문(위장관 속, 장 상피세포, 간)을 거쳐야 하며 하나씩 통과할 때마다 일부분씩 약이 제거된다. 이 세 관문을 통과하면서 약이 전신순환혈로 흡수되기 전에 제거되는 현상을 초회통과효과(first pass effect)라 한다. 그러므로 전신으로 흡수되는 약의 분율(생체이용률, bioavailability: F)은 아래 식 (1.1)과 같이 나타낸다.
\[\begin{equation} F = F_{a} \times F_{g} \times F_{h} \tag{1.1} \end{equation}\]
F는 분율(fraction)을 의미하며 Fa는 소화관 내에서 장 상피의 세포막을 투과하여 세포 내로 들어오는(Fa의 a는 absorbed를 의미함) 약의 분율로서 소화액이나 장내세균 등에 의해 약물이 흡수되기 전에 분해되어 사라지거나 소화액에 용해되는 정도, 장 상피세포를 얼마나 빠르게 투과하는지 등에 의해 결정된다. Fg는 장 상피세포 내에서 약이 대사되는 분율을 의미하며 장 상피에 많은 CYP3A4에 의한 대사가 대표적인 원인이다. 이렇게 두 번의 관문을 통과한 약은 마지막으로 간에서 다양한 효소계에 의해 대사되거나 담즙으로 배설된 후 살아남은 분율 (Fh)만이 전신순환혈에 도달하게 된다. 이 개념은 그림 1.3에 나타나 있다.
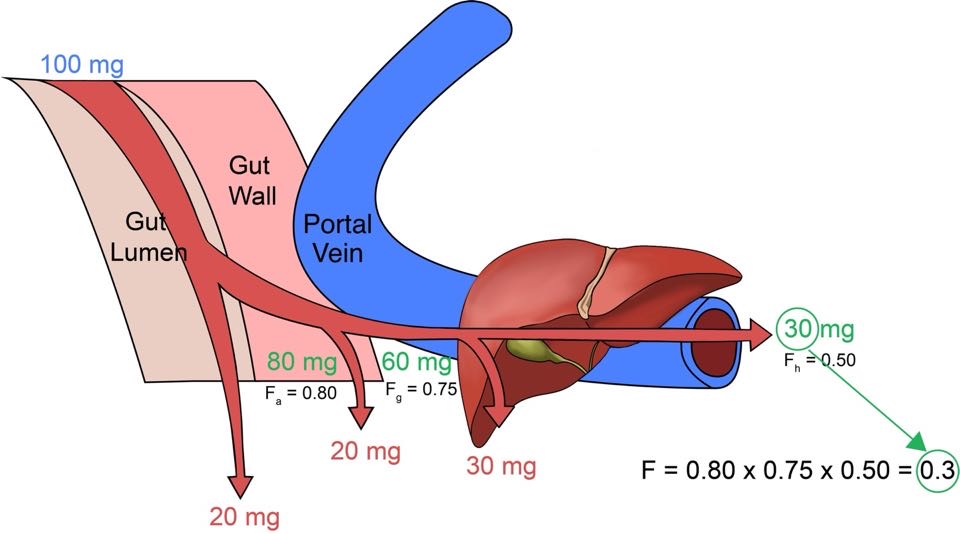
그림 1.3: F(=Fa × Fg × Fh, 생체이용률)를 구성하는 세가지 요소. 100 mg을 먹었을 때 소화관 내강과 장 상피세포, 간을 순차적으로 통과하여 전신순환혈에 도달하는 분율을 계산하는 예
1.2.2 분포(distribution)
흡수과정을 거쳐 전신순환혈에 도달한 약은 혈류를 타고 전신으로 분포하게 되는데, 이는 혈관 밖의 조직으로 약물이 확산되어 나감을 의미한다. 분자량이 작은 약일수록, 지용성이 높은 약일수록 각 조직의 세포막을 통과하기 쉬우므로 빠르게 확산되고 많은 양의 약물이 조직에 퍼지게 될 것이다. 간이나 콩팥과 같이 혈류량이 풍부한 장기로는 전신순환혈로 들어온 약이 혈류를 따라 혈관 속에 퍼지는 것과 거의 비슷한 속도로 분포가 일어나지만 그 이외의 장기들로는 시간차를 두고 분포하게 된다. 이 분포의 정도를 나타내는 파라미터가 Vd(분포용적, volume of distribution)이다.
1.2.3 대사(metabolism)와 배설(execretion)
약이란 체외에서 들어온 이물질이므로 흡수 시 초회통과를 거치면서 소장 상피세포와 간에서 대사될 뿐 아니라, 흡수가 끝난 후에도 혈류를 따라 간에서 다른 물질로 대사된 후 담즙이나 소변으로 배설되거나, 대사 없이 곧바로 배설되기도 한다. 약물의 대사과정이나 효소에 대한 지식들은 다른 책에서 얻을 수 있으므로 여기서는 다루지 않는다. 몸에서 제거되는 속도에 관여하는 파라미터가 CL(청소율, clearance)이다.
1.2.4 일차약동학(first-order kinetics)과 반감기(half-life, t1/2)
20세기 초에 혈장약물농도를 측정하는 기술들이 나오면서 정맥주입 후 측정한 약물농도는 아래 그림 1.4와 같이 시간에 따라 초기에는 급격히 감소하다가 나중에는 완만하게 떨어지는 오목한 곡선모양을 보이는 것을 알게 되었다. 로그 농도 단위로 그리면 농도 곡선은 직선으로 변하므로 ln(C) = ln(C(0))-kt와 같은 수식으로 표기할 수 있다. 여기서 C(0)란 시간 0에서의 농도이고 x축에 표기된 시간 t에 따라 기울기 -k를 가지는 직선이다. (시간의 함수로서 농도 C는 C(t)로 표기할 수 있다.) 로그 단위를 없애기 위해 이 식의 양 변에 지수를 취하면 C = C(0)·exp(-kt) 와 같이 변형되고, 다시 미분방정식의 형태로 변형하면 dC/dt = -kC 과 같은 식이 된다. 이 간단한 형태의 미분방정식이 의미하는 것은 매 순간마다 농도의 시간에 따른 변화 정도(dC/dt)가 농도의 1승(C=C1)에 비례(-kC)한다는 것이다. 시간이 갈수록 그 변화의 방향이 감소하는 쪽이므로 비례상수 k에 -가 붙는다. 이와 같이 어떤 값의 변화율이 그 값 자체(의 1승)에 비례하는 현상을 1차식(first-order kinetics)을 따른다고 한다. 우리가 아는 거의 모든 약들이 치료용량 범위에서 1차식에 따라 제거되는 것으로 알려져 있다. 경구투여 약들의 경우 소화관에서 전신순환혈로 흡수되는 속도 역시 정확하지는 않으나 편의상 1차식에 따라 흡수된다고 흔히 가정한다.
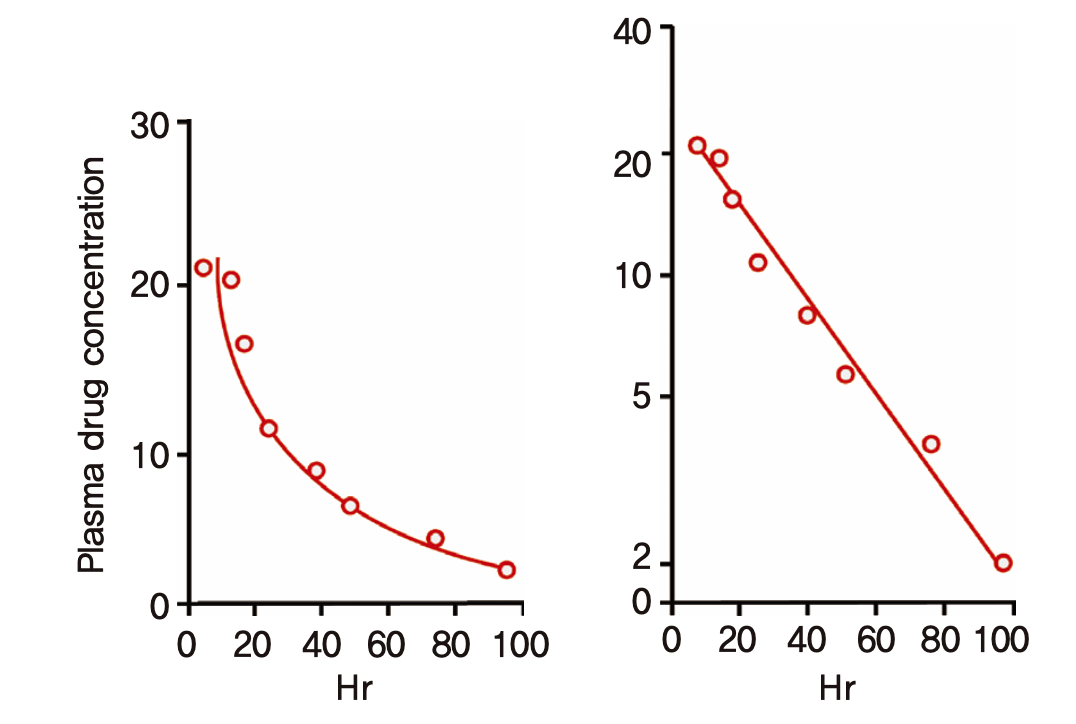
그림 1.4: 1차식에 따라 제거되는 혈장약물농도의 시간에 따른 변화
1차식에 따라 제거되는 약은 특징적으로 `반감기’(half-life, t1/2)를 가진다. 반감기는 혈장약물농도가 절반으로 줄어드는 시간이며 1차식을 따라 제거되는 약물의 경우 약물마다 제각기 일정한 반감기(t1/2)를 가진다. 보통 네 번의 반감기가 지나면 몸 안에 있는 약물이 대부분(90% 이상)이 제거되었다고 추정하고, 일정한 간격으로 연속적으로 투약할 때에도 네 번(또는 다섯 번)의 반감기가 지나면 혈장약물농도가 더 이상 높이 올라가지 않고 일정한 범위 내에서만 오르내리는 항정상태(steady state)에 도달했다고 추정한다. 반감기는 약물의 약동학적 특징을 나타내는, 쉽게 이해할 수 있는 값이지만 앞서 언급된 두 가지 주된 파라미터인 Vd와 CL의 비에 의해 결정되는 결과값이며 그 자체가 독립적인 약동학 파라미터는 아니다.
1.3 분포용적
1.3.1 약물의 분포
약물농도를 측정하던 초기에는 앞의 그림 1.4와 같이 로그 약물농도가 하나의 기울기를 가지는 직선으로 나타난다고 생각했으나, 주사기를 이용하여 정맥 내로 한꺼번에 약물을 주입하는(i.v. bolus injection) 경우 대부분 그림 1.5와 같이 주사 직후에는 급경사의 직선이, 일정 시간 경과 후 상대적으로 완만한 경사의 직선이 교차하는 형태로 나타난다는 것이 알려지게 되었다.
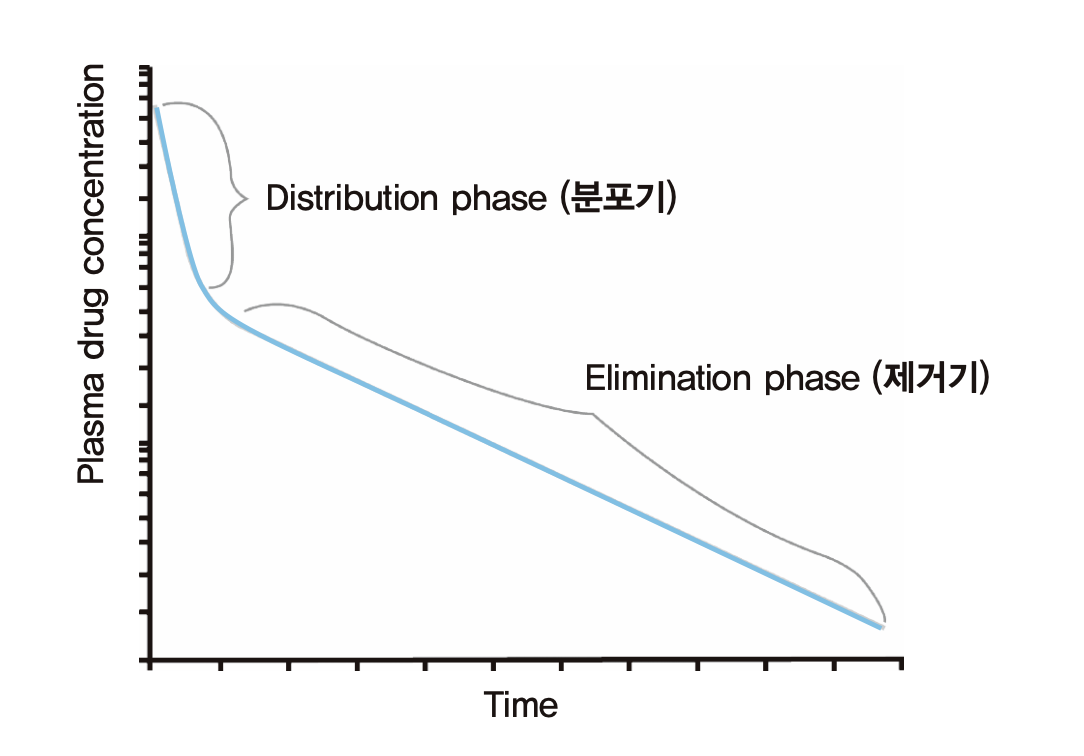
그림 1.5: 두개의 경사를 보이는 혈장약물농도 변화(i.v. bolus 주입 후 흔히 보이는 현상)
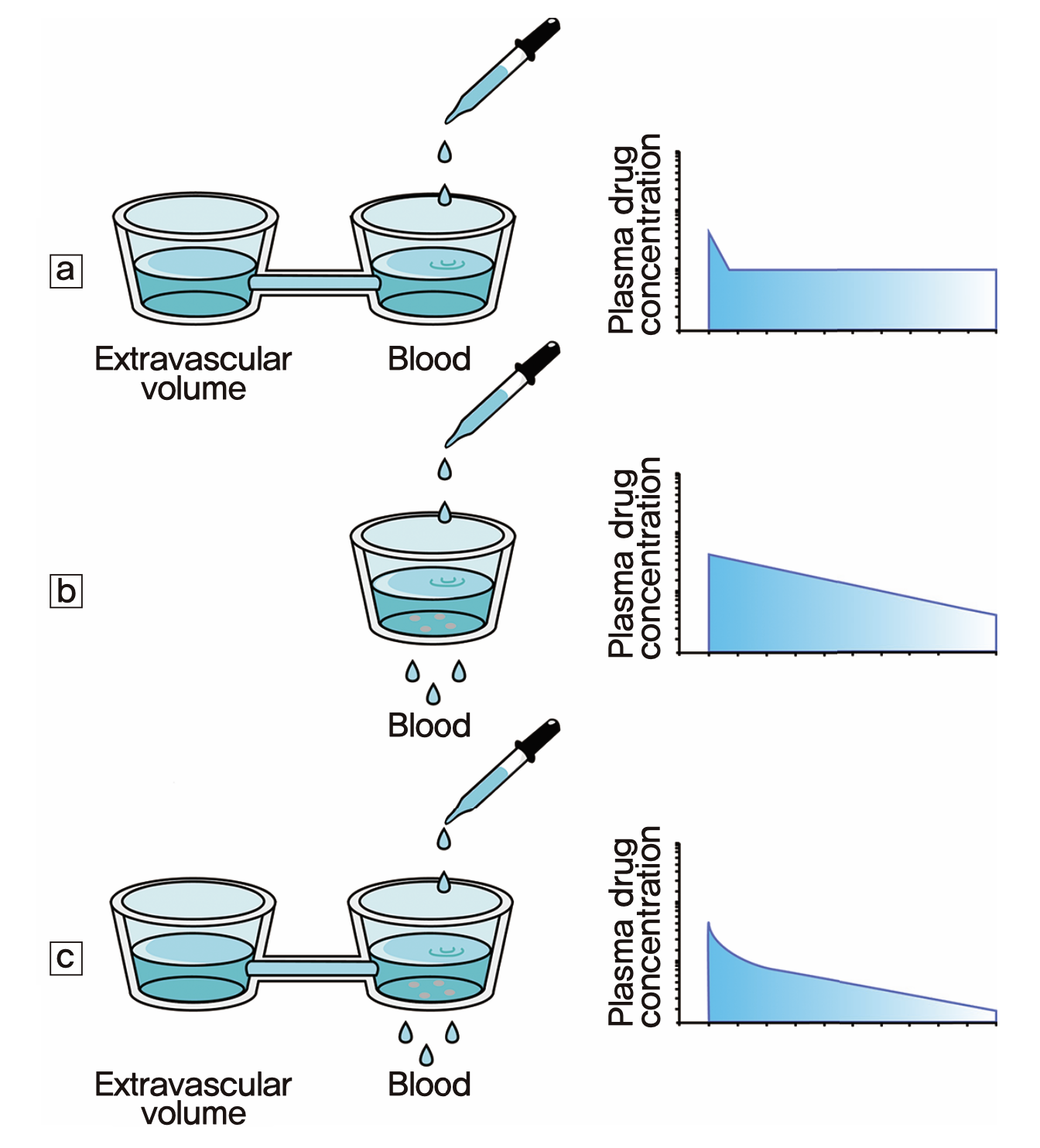
그림 1.6: 약물의 분포로 인해 두개의 기울기가 관찰되는 기전을 설명하는 모델
왜 이와 같은 현상이 나타나는지 설명하는 모델은 그림 1.6과 같은데, 사람의 몸을 물이 차 있는 하나의 비커 또는 두개의 비커가 연결된 형태로 단순화하여 가정한 것이다. 파스퇴르 피펫으로 우측 비커에 잉크를 한 방울 떨어뜨렸을 때 비커의 물 속의 잉크 농도를 시간에 따라 찍어준 것이 오른쪽의 그래프들이다. 비커 두 개가 양쪽으로 물이 통할 수 있게 연결된 상태에서 우측 비커 물 속의 잉크 농도는 맨 위의 그래프 a)와 같을 것이다. 잉크가 왼쪽 비커의 물에 골고루 확산될 때까지 급격히 떨어지고 그 이후에는 평형을 이루어서 변함이 없는 현상이다. 만약 왼쪽 비커가 없고 우측 비커 하나만 있는 상태에서 비커의 바닥에 잉크를 1차식으로 제거하는 장치가 있다고 가정하면 물 속의 잉크의 로그 농도는 가운데 그래프 b)와 같이 하나의 직선으로 나타날 것이다. 이 두 그래프를 참조하면, 두 개의 비커가 연결되어 있고 그 중 우측 비커에 잉크 제거 장치가 작동하는 상황에서 잉크농도는 그래프 c)와 같이 나타날 것이다. 잉크를 약이라고 가정하면, 우리 몸에서 보이는 혈장약물농도가 두 개의 기울기를 가진 직선으로 나타나는 원인은 이와 같이 설명되는 것이다. 즉 혈관 속이나 약물이 혈류와 거의 동시에 빠르게 퍼져서 평형이 일어나는 장기들이 오른쪽 비커에 해당하고 그 외 평형이 빨리 일어나지 않는 장기들(혈류가 낮거나, 특정 약물이 혈액-뇌 장벽 등을 통과하는데 시간이 걸리는 등)은 왼쪽 비커에 해당된다.
실제 사람의 몸에서 약이 분포하는 과정을 그림 1.7에 나타냈다. 앞 그림의 오른쪽 비커에 해당되는 것이 혈관 속(혈장), 간이나 콩팥과 같이 혈류가 많이 가고 쉽게 약물이 분포하는 장기 등이고 이를 중심구획(central compartment)라 한다. 이외에 약물이 더 늦게 분포하는 나머지 신체 부분을 말초구획(peripheral compartment)라 한다. 정맥주사 직후 혈장약물농도가 급격히 떨어지는 구간을 분포기(distribution phase)라 부른다. 그리고 두 분획 사이에 어느정도 평형이 이루어진 이후 완만한 경사로 농도가 떨어지는 그 다음 구간을 제거기(elimination phase)라 부르는데 주로 약이 몸에서 제거되는 현상에 의해 혈장약물농도가 떨어진다고 보기 때문이다. 물론 분포기에도 약이 몸에서 제거되는 기전은 작동하고 있지만, 혈장(중심구획의 일부)에서 약물 농도가 떨어지는 데 더 많이 기여하는 것은 몸에서 제거되는 기전보다 말초구획으로 이동(확산)하는 기전의 영향이 두드러진다고 보기 때문이다.
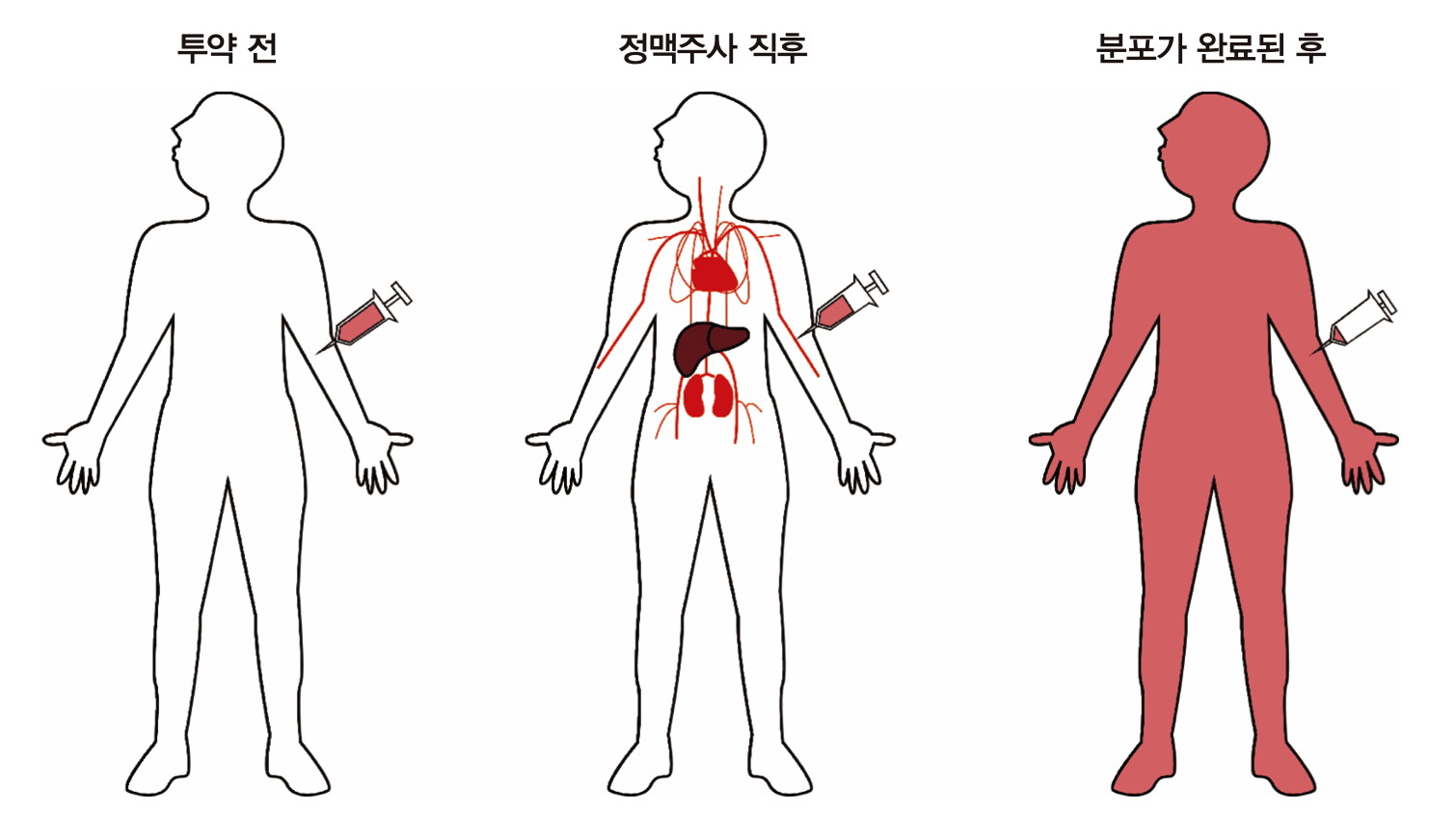
그림 1.7: 사람에서 정맥주사 후 시간에 따른 약물의 분포
1.3.2 분포용적(Vd)의 개념과 계산방법
앞서 우리는 약물이 혈관 밖의 말초조직들로 분포하는 것을 살펴보았다. 그런데, 그 분포의 정도는 약물의 물리화학적 성질에 따라 달라지며 혈장약물농도의 변화에 큰 영향을 미친다. 그림 1.8의 개념도와 같이 주사기에 검은 바둑알 모양으로 표시된 약물 12개를 담아서 bolus로 주사할 때, 약의 물성에 따라 혈장과 조직으로 분포되는 정도가 달라진다. 조직까지 분포되는데 시간이 걸리지만 개념 이해를 위해 순식간에 일어난다고 가정하자. a), b), c)의 세 경우 혈장 약물 농도는 그림에서 육면체 모양의 혈장 부피를 3L라 가정하면 a)는 1/3 개/L이고 c)는 1개/L로서 세 배의 차이가 난다. 그러나 세 경우 모두 몸 안에 존재하는 약물의 양은 12개로 동일하다. 오히려 약의 작용부위가 혈관 밖의 어떤 조직이라면 혈장농도가 가장 낮은 b)에서 조직 농도는 가장 높다는 것에 주의해야 한다. 그러므로 약물이 몸 속에서 혈장과 그 이외의 부위로 분포하는 비율을 아는 것은 중요하며, 그것을 정량적으로 나타내기 위해 Vd라는 개념을 쓰고 있다.
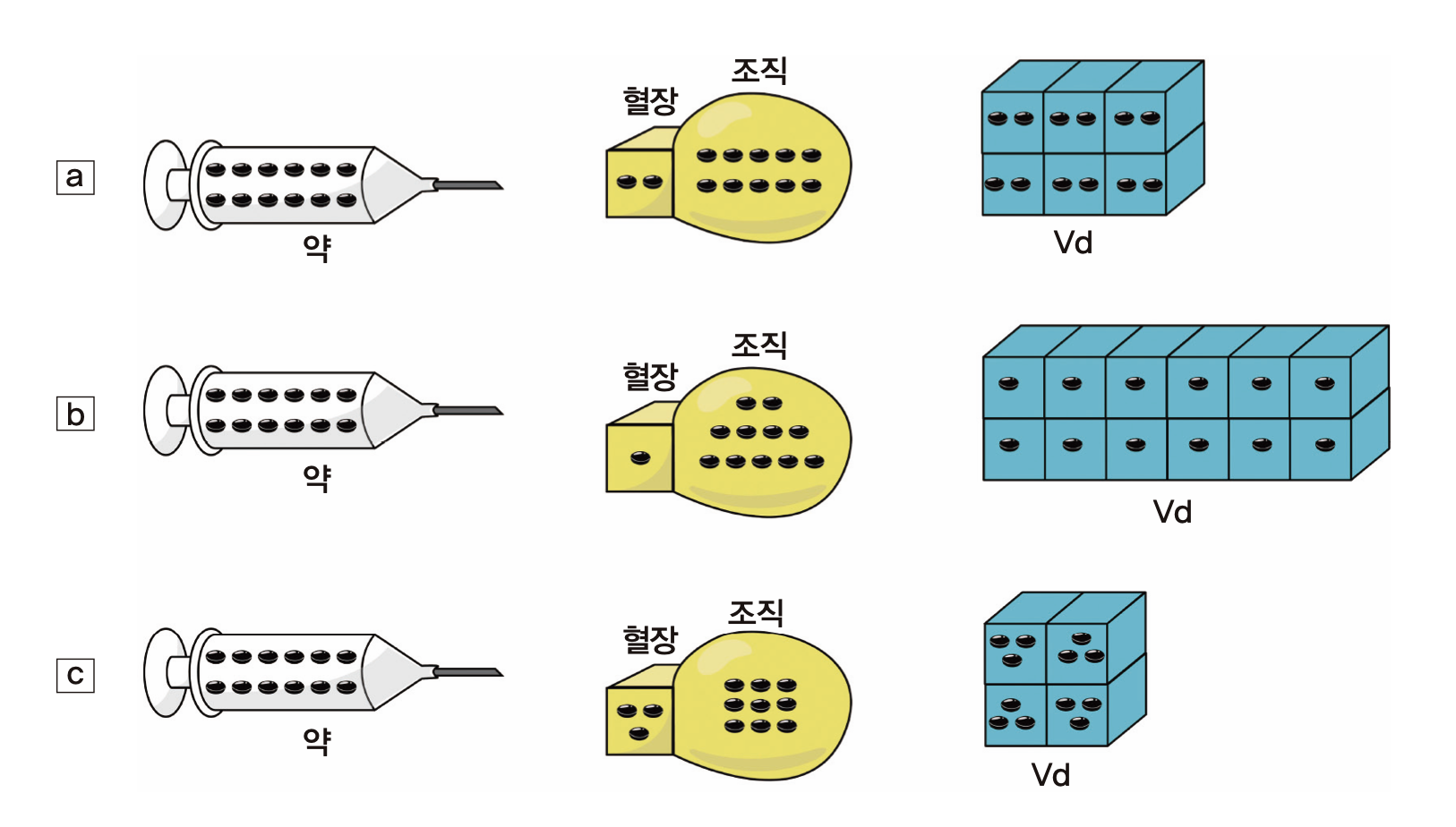
그림 1.8: 분포용적의 개념
외부에서 측정가능한 것은 혈장약물농도밖에 없는데, 12개의 약을 정맥으로 투여한 직후에 분포도 순식간에 일어났다는 가정을 하면, 몸 밖에서 안으로 들어간 약의 양 = 주사한 직후 몸 안에 있는 약의 양 = 12개 = 혈장약물농도 × ( ) 이다. 괄호 안에 들어가야 할 것은 무엇인가? 예컨데 a)의 경우라면 혈장약물농도는 2/3 개/L 이므로 18 L를 곱해야 12개가 나올 것이다. 18이란 혈장의 부피(3 L)에 6배를 곱해준 값이다. b)의 경우는 혈장 부피의 12배(36 L), c)의 경우는 4배(12 L)를 곱해주면 몸 안에 존재하는 약물의 양 12개를 얻을 수 있다. 즉 위의 괄호 안에 들어가는 부피를 혈장농도에 곱해주면 몸 안에 존재하는 약의 총량을 계산할 수 있으며, a)와 같이 혈장보다 조직에 상대적으로 많은 양의 약이 분포하는 경우 ( ) 속에 들어가야 할 값은 커지고, c)와 같이 반대의 경우 작아짐을 알 수 있다. 이 때 ( ) 속에 넣어주어야 할 값이 Vd가 되는 것이다. 다시 말하면 몸 안에 존재하는 약물의 총량을 혈장농도와 관련하여 표현하고자 할 때 혈장농도에 곱해주어야 하는 부피 값이 Vd이며, 그 크기는 약이 혈장보다 조직에 더 많이 녹아 들어가 있을수록 커진다. 실제 시판되고 있는 약들 중 그림 1.8에서 예시한 것처럼 Vd가 혈장부피의 3배-12배(9 L ~ 36 L) 정도에 그치는 약은 드물며, 조직 표면에 결합하거나 세포막을 투과할 수 있는 많은 약들이 이보다 더 큰 Vd를 나타낸다.
1.3.3 Vd와 약의 물리화학적 특성의 관계
혈관 밖의 조직으로 약이 분포하는 경향이 클수록 Vd가 커지므로 분자량이 작은 경우 혈관 외부로의 이동은 물론 세포막 통과도 쉬우므로 Vd가 클 것이다.
반대로 분자량이 150 kD에 달하는 단클론항체 등은 주사한 직후에 Vd를 측정한다면 혈장용적과 같은 값으로 나올 수 있다. (항체의약품은 소분자의약품과는 달리 혈관 내피세포를 투과하는 별도의 기전이 존재하나 혈관 밖으로의 분포가 이루어지는 것이 소분자 의약품에 비해 매우 느리다.)
분자량이 작은 약들의 경우 Vd가 커지는 경향은 있지만 약물 분자의 극성도 영향을 미친다. 그림 1.9의 mannitol과 같이 -OH 기로 인한 극성이 큰 물질은 세포막 안으로 들어가지 못하므로 그 Vd는 세포외액의 부피(체중 60 kg인 성인에서 14 L)와 유사함이 알려져 있다.
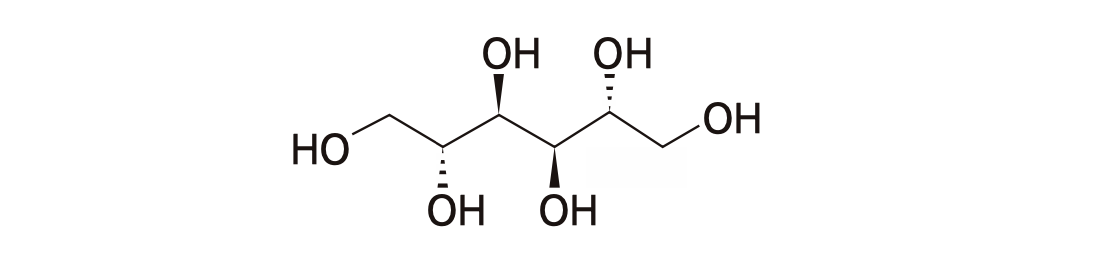
그림 1.9: 분자량은 작지만 극성으로 인해 Vd가 작은 mannitol의 분자구조.
물에 잘 녹지만 분자량이 작고 극성이 높지 않아서 세포막을 잘 투과하는 에탄올의 경우, Vd는 인체의 총 수분함량(체중 60 kg인 성인에서 42 L) 과 유사하다. 반면, 지용성이 높은 물질들의 경우 세포막을 쉽게 투과함은 물론, 세포 내의 수분에 한정되지 않고 지질 속으로 그 용해도만큼 녹아 들어갈 수 있으므로 매우 큰 Vd를 나타낼 수 있다(예, propofol: 500 L). 지용성 뿐 아니라 특정 조직에 분포하는 성질 또한 Vd에 영향을 주는데, 골격근 조직 표면에 결합함으로써 혈장보다 조직에 100배 가량 높은 농도로 분포하는 digoxin의 Vd는 500 L 내외임이 알려져 있다. 그러므로 이와 같이 큰 Vd 값은 사람 몸에서 얻을 수 있는 생리적인 부피와는 무관하고 단지 약물이 혈관 밖에 상대적으로 더 많이 분포한다는 것을 의미한다.
약이 효과를 나타내는 타겟 세포나 조직에 얼마나 분포하는지는 단순히 약동학 뿐 아니라 유효용량이나 유효혈장농도를 결정함에 있어서도 매우 중요한 정보이다. 실제 환자에서 약물의 Vd는 혈장의 용적, 조직의 용적이 큰 사람일수록 커지므로(즉 체중이 클수록 커지므로) 보통 L/kg의 단위로 표기하게 된다.
1.3.4 Vd와 혈장단백결합률의 관계
대부분의 약동학 교과서들에서 전혀 언급되지 않는 내용이지만, 혈장단백결합률과 약물의 분포의 관계에 관해서 많은 이들이 오해하고 있는 부분이 있어서 이 장에서는 별도의 섹션으로 다루었다. 약물의 혈장단백 결합은 van der Waals 힘이나 수소결합 등에 의한 소수성 부위들의 상호작용이나 정전기적 상호작용에 의한 미약한 결합이다. 여기서 혈장단백질이란 주로 알부민이나 α1-acid glycoprotein이다. 혈장에서는 단백결합약물과 비결합약물의 분율이 약물마다 고유한 값을 가지는데, 예컨데 0에서 1사이의 fu (unbound fraction, 비결합분율) 값으로써 나타낸다. 즉 fu가 1이면 전혀 혈장단백질에 약물이 결합하지 않는 것이고 0.1이면 90%의 약물이 혈장단백질에 결합되어 있고 10%만이 비결합형으로 존재하는 것이다. 이들 혈장단백질은 분자량이 커서 혈관 밖으로 쉽게 빠져나오지 못하므로 혈장단백질에 결합한 약물들도 따라서 혈관 밖으로 빠져나오지 못하여, 혈장단백 결합률이 높은 경우 Vd가 작을 것이라고 추정할 수 있으나 실제 관찰되는 현상은 그렇지 않다.
혈장단백질에 결합하지 않은 약(unbound drug, free drug)은 분포의 평형이 이루어졌을 때 혈장과 조직에서의 농도가 동일하다고 간주한다.(이는 지나치게 단순화시킨 주장이며 이것이 성립하기 위해서는 여러가지 조건이 붙지만 약동학을 처음 배우는 수준에서는 그냥 받아들이자.) 혈장단백질에 결합하지 않은 약물이 조직 내로 이동하면 그 조직내에 존재하는 단백질 등에 혈장단백 결합과 마찬가지의 기전으로 결합하게 되고 조직마다 약물에 따라 고유한 조직결합율을 나타내게 된다. 조직결합율이 높거나, 조직 내에 약물의 용해도가 높은 경우에는 혈장에 있던 비결합 약물들이 대거 조직 내로 확산되어 이동하게 된다. 그러면 혈장에서는 약물마다의 고유한 특성이라 할 수 있는 본래의 fu 값을 유지하기 위해 단백결합약물들이 대거 떨어져 나와서 조직으로 이동한 비결합약물의 빈 자리를 채울 것이다. (그림 1.10)
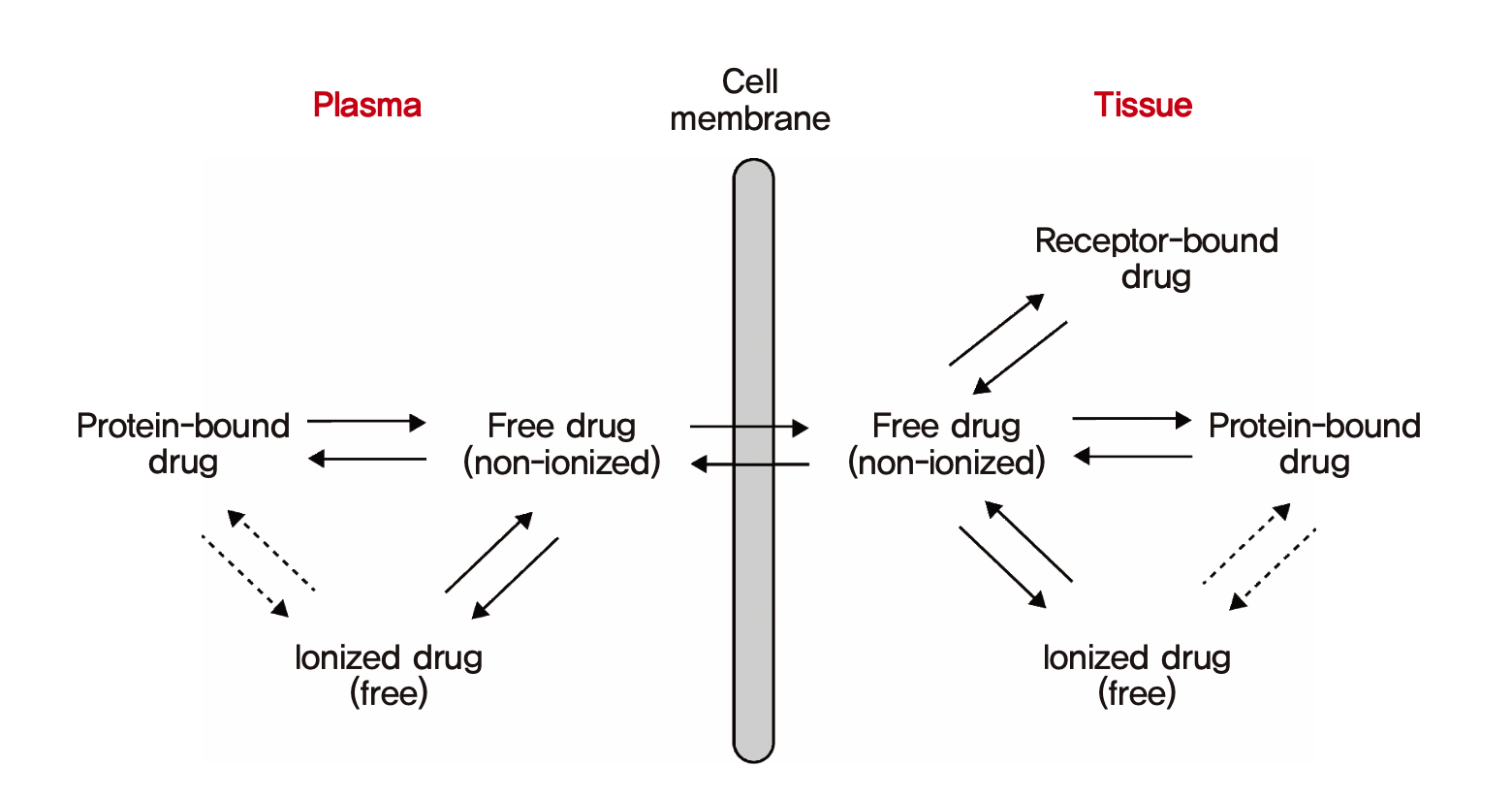
그림 1.10: 혈장단백결합과 조직결합, 약물의 분포의 관계 (모세혈관과 세포외액, 혈장의 구분은 생략되어 있음)
결론적으로 아무리 혈장단백결합율(=1- fu)이 높아도 조직결합율이 높거나 조직에 대한 용해도가 높은 경우(지용성이 높은 약 등)에는 혈관 밖의 조직들로 약물이 퍼져 나가는 것을 막을 수가 없어서 혈장단백결합율만 가지고 Vd가 크거나 작을 것이라고 미리 단정하는 것은 무리이다. 그림 1.11은 알려진 670가지 소분자 의약품의 약동학 파라미터들을 정리한 표(Obach, Lombardo, and Waters 2008)를 바탕으로 그려본 fu와 Vd의 관계로서 어떤 경향성을 찾을 수 없다는 것을 알 수 있다. 다만 동일한 약에서, 질병 등으로 인해 혈장단백질 결합율이 감소하는 경우(간기능 이상 등으로 혈장단백질 자체가 감소하거나 요독증으로 인해 혈장단백질과 약물의 결합부위를 두고 약물과 경쟁하는 혈중 노폐물 등의 증가)에는 Vd가 건강인에 비해 증가함이 알려져 있다. 물리화학적 특성과 화학 구조가 유사한 같은 계열의 약물들 간에서도 혈장단백질 결합율이 높으면 분포용적이 작은 경향성이 발견된다. 그러나 이런 특수한 상황을 제외하면 일반적으로 혈장단백질 결합율과 분포용적의 상관성은 찾아보기 어렵다.
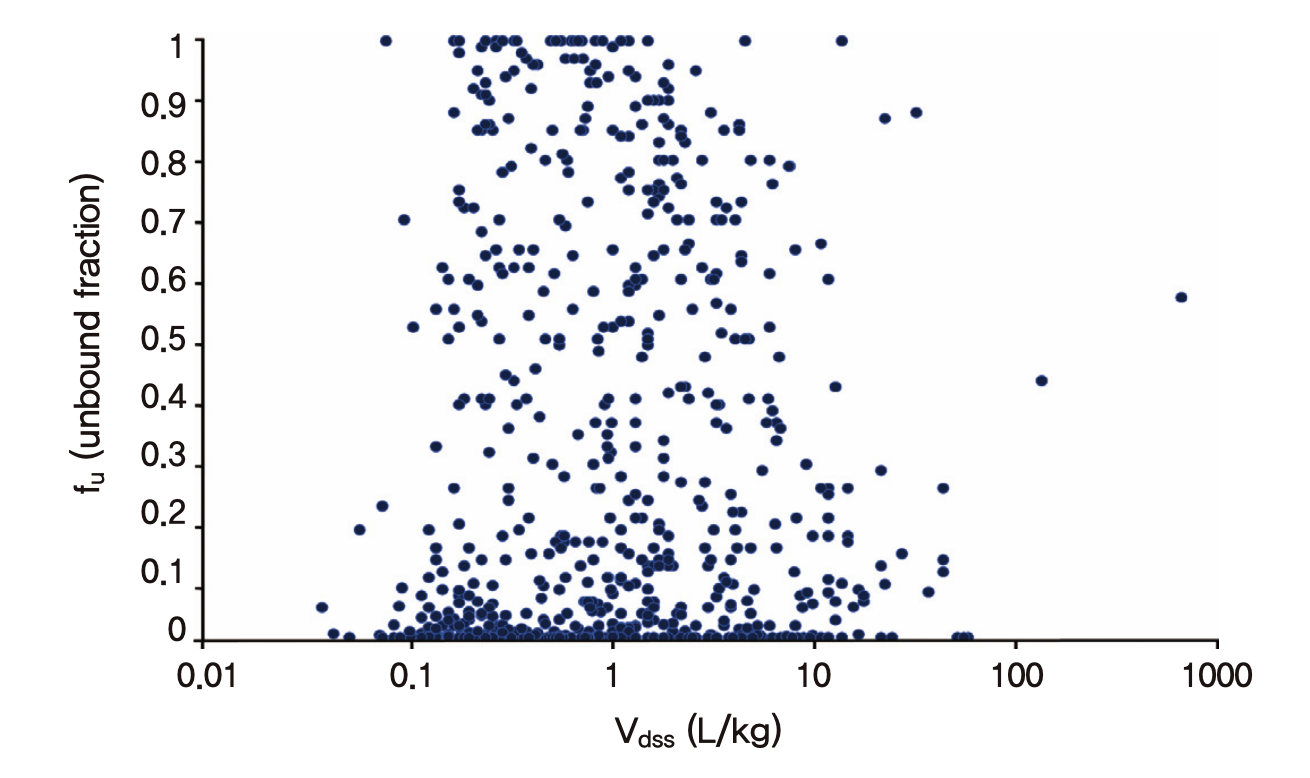
그림 1.11: 알려진 670가지 소분자 의약품의 약동학 파라미터(Obach, Lombardo, and Waters 2008)를 이용하여 그려 본 Vd(Vdss)와 fu의 산점도: fu만으로 Vd를 예측하는 것은 어려움을 알 수 있다.
1.3.5 Vd가 약동학에 미치는 영향
Vd가 클수록 같은 양을 투여해도 약물의 최고 혈장농도 (Cmax)는 낮을 것이다. 또 Vd는 1.4절에서 설명할 CL와 함께 약의 반감기를 결정하는 두가지 파라미터 중 하나이다. 같은 CL를 가진 약물들이라 해도 Vd가 다르면, Vd가 더 큰 약일수록 반감기도 늘어난다. 약물의 흡수를 나타내는 것은 ka와 F지만, 일단 소화관내의 약이 모두 전신순환혈로 들어온 이후에 약의 혈장농도를 결정하는 것은 Vd와 CL 뿐이다.
1.4 청소율(CL)의 개념과 적용
1.4.1 청소율의 이해
몸이 약을 제거하는 능력을 시간당 제거하는 약물의 양(mg/h 와 같은 단위)으로 나타내는 대신, 시간당 그 약이 녹아 있던 혈장이나 혈액을 얼마나 청소해 주었는지(혈액이나 혈장의 부피)로 나타내는 것(L/h 등의 단위)이 청소율(CL)이다. 즉 용매(혈장)에 녹아 있는 용질(약)을 제거하는 몸의 능력을 용질보다는 용매를 중심으로 표현하는 것이다. 왜 번거롭게 이런 식으로 표현하는가? 그것은 시간당 제거되는 약의 양이라는 단위로 표현하면 약의 혈장 농도가 높을 때는 제거 능력이 상대적으로 크다가 농도가 떨어지면 줄어든 값이 되어 일정한 값으로 표현하는 것이 불가능한 반면, 시간당 청소되는 혈액이나 혈장의 부피는 같은 사람에서 약물별로 그 약물의 혈장농도와 무관하게 고정불변의 값을 나타내기 때문이다. 이러한 고정불변의 CL가 있기 때문에 약물이 1차식에 의해 제거되고 일정한 반감기를 나타내지만, 처음 배우는 사람은 이 개념을 이해하는 것이 어렵다. 이해를 돕기 위해 약을 제거하는 장기(간이나 콩팥)를 진공청소기로, 혈장을 공기로, 약물을 먼지로 비유하여 설명하고자 한다. 이 비유적 사례를 통해 왜 CL를 장기의 약물제거 능력을 나타내는 파라미터로 쓰는지 이해할 수 있을 것이다. (Yim 2017)
사례
당신은 공기 중에 먼지가 100 ng/L의 농도로 존재하는 방에 있다. 이 방의 공기를 새로 구입한 진공청소기를 이용해서 청소하고자 한다. 스위치를 켜고 노즐을 허공을 향하게 하여 30분간 진공청소기를 돌리고 나서 청소기를 분해하여 필터(집진주머니)에 걸린 먼지의 양을 저울로 측정해 보니 200 ng 이였다. 그러면
1. 문: 이 청소기가 청소한 공기의 부피는 얼마인가?
답: 30분에 200 ng의 먼지를 제거했으므로 2 L의 공기를 청소한 것이다.2. 문: 시간당 청소한 공기의 부피는?
답: 30분동안 2 L의 공기를 청소했으므로 4 L/h의 공기를 청소했다(청소기의 성능을 CL로 나타냈다.).3. 문: 시간당 제거한 먼지의 양은?
답: elimination rate = CL × conc = 4 L/h × 100 ng/L = 400 ng4. 문: 세시간동안 청소기를 돌린다면 제거한 먼지의 양은?
답: Amount eliminated = CL × conc × Δt(duration) = 4 L/h × 100 ng/L × 3 h = 1200 ng5. 문: 이 청소기를 공기 중에 먼지가 50 ng/L의 농도로 존재하는 옆방에 가져가서 작동시킨다면 시간당 제거한 먼지의 양은?
답: CL × conc = 4 L/h × 50 ng/L = 200 ng 청소기의 고유한 성능인 CL는 그대로이지만, 먼지의 농도가 절반인 방에서 시간당 제거한 먼지의 양은 첫 번째 방의 절반밖에 안된다. 만약 공기 중에 먼지가 하나도 없다면 시간당 제거한 먼지의 양은 0 일 것이다! 따라서 시간당 제거한 먼지의 양으로 청소기의 성능을 표현하는 것은 옳지 않고, CL로 표현해야 한다.6. 문: 위의 사례에 제시된 질문 1)에서 5)를 풀 때의 가정은 방이 아주 넓어서 청소기로 먼지를 몇시간 빨아들였다고 해서 방안 공기 중의 먼지 농도는 전혀 변치 않는다는 것이었다. 그러나 실제 사람의 몸에서 간이나 콩팥이 약을 제거하면 혈장약물농도는 지속적으로 떨어지는 것을 관찰할 수 있다. 이런 상황에서 CL를 어떻게 구하겠는가?
답: 아주 짧은 시간(Δt)동안에는 약물농도(시간의 함수로 C(t)로 표기)의 변화가 무시할 만큼 작으므로 Amount eliminated = CL × C(t) × Δt 이다. 0에서 ∞ 시간의 간격동안 제거된 약물의 양은 몸 안에 들어온 약물의 양과 같고, 이는 정맥주사(F=1.0)로 투여한 용량과 같다. 이는 또한 0에서 ∞ 시간을 극히 작은 간격 Δt들로 쪼개서 그 간격마다 제거된 약물의 양을 모두 합해준 것과 같다. 따라서 아래와 같은 식이 성립한다.
\[ Dose = \lim_{\Delta t \rightarrow 0}{\sum_{}^{}{CL \cdot C\left( t \right) \cdot \Delta t = CL\int_{0}^{\infty}{C\left( t \right)dt}}} \]
결국 외부에서 정맥주사로 투여한 용량을 알고, 시간에 따라 변화하는 혈장약물농도 곡선하 면적(AUC: area under the concentration-time curve =\(\int_{0}^{\infty}{C\left( t \right){dt}}\))을 구하여 CL를 계산하는 것이 흔히 쓰는 방법이다. AUC는 적분계산을 하지 않더라도, 채혈시간과 측정된 혈장약물농도 값으로 그린 곡선의 면적을 사다리꼴의 공식 등으로 계산하여 근사적으로 구할 수 있는데 그 방법은 NCA(non-compartmental analysis, 비구획분석)장에서 다룬다.1.4.2 청소율과 k, t1/2의 관계
앞서 약의 분포를 나타내는 파라미터가 Vd이고 제거능력을 나타내는 파라미터는 CL임을 배웠다. 그러면 그림 1.4에 나타난 것과 같은 직선의 경사에 해당하는 약물의 제거속도상수(k 또는 ke로 표기: e는 elimination을 의미함)는 k = CL/Vd의 관계를 따른다. 그 이유는 아래와 같이 설명할 수 있다. 제거속도상수 k는 뭔가가 사라지는 속도(rate)를 표기하기 위해 쓰는 비례상수라는 의미인데 혈장약물농도가 떨어지는 속도(dC/dt)의 경우 dC/dt = –k·C로 정의되고, 체내에 남아있는 약물의 총량(A: amount)이 줄어드는 속도(dA/dt)를 나타내고자 할 때는 dA/dt = -k·A와 같이 표기할 수 있을 것이다. 그런데 앞부분에서 나온 분포용적의 개념으로 추론하면 어느 순간 체내에 존재하는 약물의 양 A = C·Vd이므로 -k·A = -k·C·Vd 이고 또 CL의 정의로 나온 제거 속도는 CL·C이므로, -k·C·Vd = CL·C가 된다. 그러므로 결국 k = CL/Vd이다. (감소를 의미하는 - 부호 생략) k의 단위는 시간의 역수(1/h)가 된다.
혈장약물농도의 반감기는 일차약동학의 또다른 수식인 C(t) = C(0)×exp(-kt)에서, 시간의 함수인 C(t)가 처음 농도 C(0)의 1/2로 줄어드는데 걸리는 시간으로 구할 수 있다. 즉 C(t) = 1/2·C(0) = C(0)·exp(-kt)에서 양 변에 자연로그를 취하고 풀어주면 ln(½) + ln(C(0)) = ln(C(0)) – kt이고 t (=t1/2) = - ln(½)/k = ln(2)/k = 0.693/k이다. 즉 t1/2는 k에 반비례하고 k = CL/Vd이므로 CL에 반비례하고 Vd에는 비례한다. 예컨데, CL는 몸에서 약을 제거하는 능력을 나타내는 값인데, 똑같은 CL를 가진 서로 다른 두 약의 Vd가 두 배 차이 난다면 Vd가 큰 약의 반감기가 다른 약의 반감기의 두 배가 될 것이다. 직관적으로 이해하기 위해서 분포모델을 생각해 본다면, 시간당 약이 녹아있는 용매인 혈장을 간에서 청소하는 능력(CL)은 A와 B라는 두 약에서 똑같지만, 혈관 외 조직에 약이 녹아있는 정도(농도, 양)가 A에 비해 B가 두 배 크다고 하자. 즉, B의 Vd가 A보다 2배 크다면, 혈장이 청소되어 약물농도가 낮아지려 할 때, B는 A에 비해 조직에서 혈장으로 약이 그만큼 더 많이 이동하여 혈장에서 사라진 약물 분자들의 빈 자리를 더 빨리 채울 것이다. 따라서 Vd가 크면 클수록 혈장약물농도를 빨리 떨어뜨리는 것이 힘들다(반감기가 길다)는 것을 알 수 있다.
1.4.3 PK 파라미터들은 약마다, 사람마다 다르다
지금까지 열거된 PK 파라미터들인 ka, F, CL, Vd는 모두 각 약물마다 제각기 다른 값들을 가지고 있다. 이들 파라미터 값들이 약물분자들의 물리화학적 특징에 따라 일정한 경향성을 나타내기 때문에 그러하다. 물론 이것을 사람들에서 약동학 시험을 통하여 환산한 파라미터 값들의 평균이 약마다 제각기 다르다는 뜻이다. 또, 같은 약이라 해도 사람 각자에서의 PK 파라미터 값들은 제각기 다른 값을 나타낸다. 예컨데 소변으로 배설되는 약은 신기능이 떨어지는 노인에서는 CL이 젊은이보다 낮게 나올 것이고, 지방조직으로 잘 분포되는 지용성 높은 약은 체중당 Vd가 비만한 사람에서 마른 사람보다 높게 나올 것이다. 결국 동일한 성분의 약에 대해서도 사람마다 체중, BMI, 장기 기능, 질병상태 등이 조금씩 다르기 때문에 이런 개인간의 차이가 발생하는 것이다. 각자에서 측정된 PK 파라미터들은 그 사람의 신체상태, 건강상태가 급격히 변하지 않는 한 PK 시험을 반복하여 구해봐도 거의 같은 값을 나타낼 것이다. 즉 개개인의 PK 파라미터는 약의 물리화학적 특성과 개인의 신체적 특성(체중, BMI, 장기 기능, 질병상태 등)이 상호작용하여 정해지는 것이라 할 수 있다.
1.4.4 CL와 Vd의 관계에 관한 고찰
1.4.4.1 CL와 Vd의 독립성
우리는 앞에서 CL와 Vd의 관계가 k =CL/Vd라는 수식으로 나타낼 수 있음을 보았다. 이 식은 제거속도상수 k가 CL와 Vd에 의해 결정된다는 것을 의미하는 것이다. 이 식을 변형하면 CL=kVd 또는 Vd=CL/k로도 표기할 수 있으나 그렇다고 해서 Vd값이 커지면 CL이 비례해서 커진다거나(CL=kVd), CL값이 커지면 Vd가 비례해서 커진다는(Vd=CL/k) 식으로 해석하는 것은 잘못된 해석이다. 다시 강조하지만 CL와 Vd는 k에 의해 바뀌는 값이 아니라, k는 종속변수로서 독립변수들인 CL와 Vd에 의해 변하는 값이라는 것이다. 그러면 CL와 Vd는 어떤 관계인가? CL는 주로 몸이 약을 대사시키거나 배설시키는 능력에 의해 정해지고 Vd는 주로 약의 물리화학적 특성에 의해서 정해지는 파라미터이므로 본질적으로는 연관성이 없다. 예컨데 지용성이 높거나 분자량이 작아서 Vd가 큰 약이라고 해서 간에서 대사가 더 빨리 되거나 소변으로 많이 배설된다는 보장은 없다. 높은 지용성, 작은 분자량을 가진 약은 간 세포막을 쉽게 통과하여 세포 속으로 잘 들어가지만, 세포 속에 많이, 빨리 들어가는 것과 CYP 효소들에 의해 빨리 대사되는 것과는 무관하다는 것이다(대사속도에는 CYP효소에 의해 분해되는 정도를 좌우하는 약물 분자의 화학 구조들이 지용성이나 분자량보다 훨씬 중요하다). 또 지용성이 높으면 세뇨관을 통한 재흡수율이 높아져 콩팥에 의한 CL는 오히려 떨어질 것이다. 그러므로 670개 성분에서 보고된 CL와 Vd의 산점도(scatterplot)을 그려보면 아무런 경향성도 찾을 수 없다. (그림 1.12)
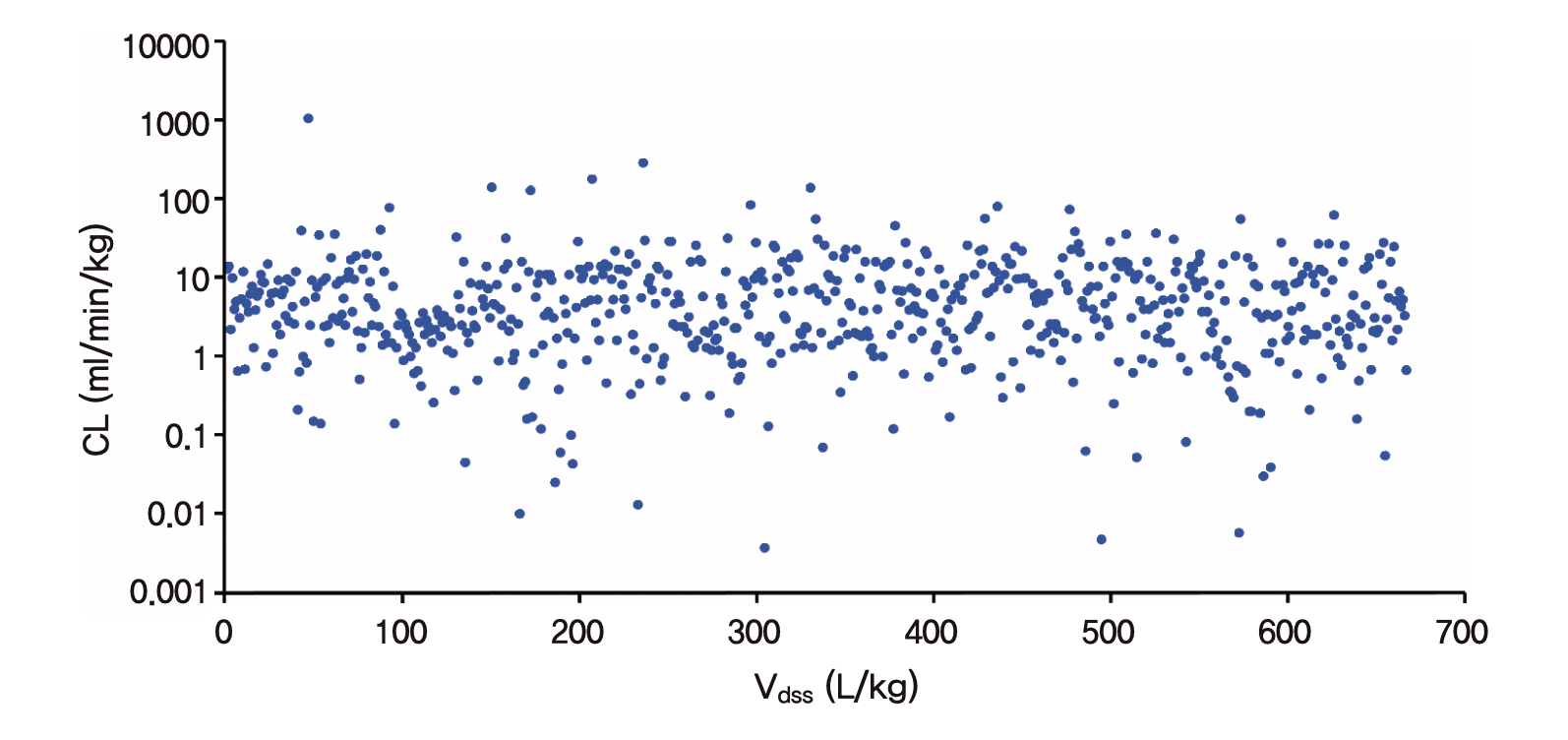
그림 1.12: 670가지 약물에서 보고된 CL와 Vd로써 그려 본 산점도 (Obach, Lombardo, and Waters 2008): CL과 Vd는 아무 관계도 없음을 알 수 있다.
1.4.4.2 개인별 CL와 Vd의 상관성
다만, 약의 물리화학적 특성에 무관하게, 한 가지 성분의 약을 골라서 사람들에게 투여하여 얻은 각자의 CL와 Vd를 비교해 본다면, Vd가 작은 사람이라면 그 사람의 CL도 작은 경향이 있고, Vd가 큰 사람에서는 그 사람의 CL도 큰 경향이 있다. 모든 약의 Vd는 체중이 클수록 커지는 경향이 있다. CL의 경우는 콩팥과 간에 의한 대사속도를 반영하는데 콩팥의 약물제거 속도를 결정하는 사구체여과율(GFR)은 이미 체중과 비례관계가 있음이 잘 알려져 있다. 간에서 대사되는 경우라 해도 간 대사 효소의 총량이 간 세포의 숫자와 비례할 것이므로 CL는 간의 무게와 비례하고, 간의 무게는 체중이 클수록 커지는 경향이 있으므로 체중이 크면 간에 의한 CL도 커지는 경향이 보이기도 한다. 물론 체중보다 각자의 간 대사 효소의 유전적 대사능이 CL에 미치는 영향이 더 클 경우는 상관관계가 미약할 수도 있다. 어떤 약이건 환자 각자에서 얻어진 CL와 Vd는 양의 상관관계가 있다. 이런 체중과 CL의 상관성을 무시하고 집단약동학(population PK) 모델을 만들면 시뮬레이션을 수행할 때 현실과 동떨어진 가상데이터(예컨데 어떤 약에 대한 Vd가 아주 크면서 CL는 아주 작은 가상환자)를 만들어낼 위험이 있다.
1.5 다회투여(multiple dose)의 약동학
첫 번 투약하고 24 시간이 지난 후 약이 몸 안에서 완전히 제거되지 않았는데도 다음 번 투약을 하고, 이것을 일정한 시간 간격으로 반복하면, 혈장약물농도가 한없이 올라갈 것 같지만, 1차식에 의해 제거되는 약의 농도는 그렇지 않고 일정한 범위 안에서 머물게 되어 있다. 대부분의 약들의 용량용법은 일정 간격(24 시간 등)으로 여러 번 투여하여 약물농도가 일정 범위 안에서 올라가고 내려감을 반복할 때 약효가 가장 잘 나타나도록 설계되어 있다.
일정 간격으로 계속 투약하고 시간이 충분히 경과하면 농도가 이렇게 일정한 범위(항정상태, steady state) 안에 머물게 되는 것은 1차식에 의한 제거라는 특징 때문이다(1차식에 의해 제거되지 않는 약, 예컨데 Michelis-Menten kinetics 를 따르는 약은 이렇지 않은데, 비선형약동학을 따른다고 하며, 이후에 설명한다.)
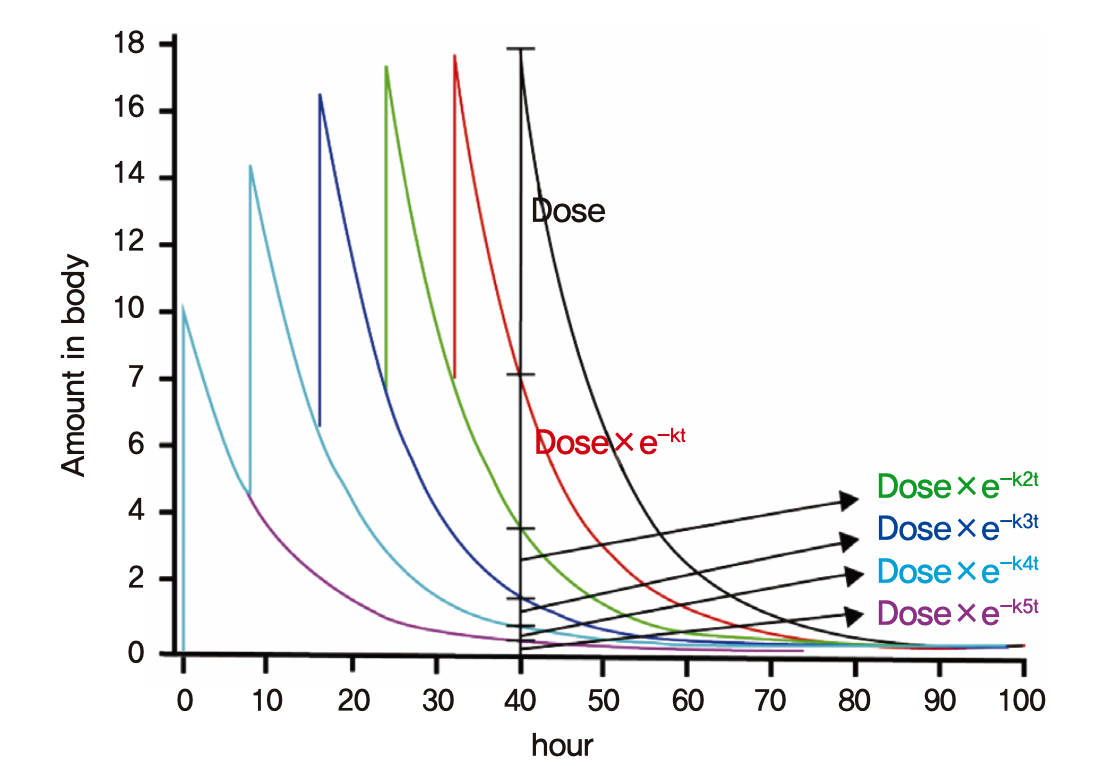
그림 1.13: 정맥주사(i.v. bolus) 6회 투약 후 체내 약물의 양
그림 1.13의 예와 같이 일정시간(τ) 간격으로 정맥주사(i.v bolus)로 6번 투약을 한 직후의 몸 안에 들어있는 약물의 양은 이전에 여러 번 투여된 약들이 몸에 남아있는 양을 모두 합친 것(중첩된 것)과 같을 것이다. 방금 혈관에 주입된 양(Dose) + τ 시간 전에 들어와서 남아있는 양(Dose × e-kτ) + 2τ 시간 전에 들어와서 남아있는 양(Dose × e-k2τ) + 3τ 시간 전에 들어와서 남아있는 양(Dose × e-k3τ) + 4τ 시간 전에 들어와서 남아있는 양(Dose × e-k4τ) + 5τ 시간 전에 들어와서(첫 번째 투여) 남아있는 양(Dose × e-k5τ)이 그 값이다. 혈장농도 역시 약물의 양과 같은 양상을 보일 것이다(몸안에 존재하는 약의 총량을 Vd로 나누기만 한 값이므로). 그림에서와 같이 최고농도(Cmax)가 커지는 정도는 투여를 거듭할수록 약해지면서 언젠가는 더 이상 올라가지 않는 양상을 보이게 될 것이다. 만약 n번 투약을 했다면 식 (1.2)로 표기될 수 있고 이는 초항이 Dose/Vd이고 공비가 e-kτ인 등비수열의 n항까지의 합을 구하는 것과 같은 원리이다. 공식에 의해 n번째 용량에서의 Cmax = Dose/Vd × (1- e-knτ)/(1- e-kτ)로 나타난다.
\[\begin{equation} \begin{split} C_{max} &= \frac{Dose}{V_{d}} \times \left( 1 + e^{- k\tau} + e^{- k2\tau} + e^{- k\tau 3} + \ldots + e^{- k\left( n - 1 \right)\tau} \right) \\ &= \frac{Dose}{V_{d}} \times \frac{1 - e^{- kn\tau}}{1 - e^{- k\tau}} \end{split} \tag{1.2} \end{equation}\]
여러 번 투여할수록(n이 커질수록) e-knτ는 작아져서, 이론적으로 무한한 시간이 지나서, 무한번(n = ∞) 투여한 상황이 되면 e-knτ = 0이 되고 그 때의 농도는 비로소 더 이상 증가하지 않는 항정상태의 농도가 될 것이다(식 (1.3)).
\[\begin{equation} {C_{max\_ ss} = \lim_{n \rightarrow \infty}}{\frac{{Dose}}{V_{d}} \times \frac{1 - e^{- kn\tau}}{1 - e^{- k\tau}} = \frac{{Dose}}{V_{d}} \times \frac{1}{1 - e^{- k\tau}}} \tag{1.3} \end{equation}\]
그러나 현실에서는 무한대라는 시간을 적용하는 것이 불가능하고 대신 앞서 나온 Cmax = Dose/Vd × (1- e-knτ)/( 1- e-kτ) 수식의 분자 부분의 e-knτ가 0에 대략 비슷할 정도로 충분히 작아지는 시간을 추산하여 쓴다. e-knτ 식에서 지수부분(-knτ)은 제거속도상수 k와 경과시간 nτ의 곱으로 이루어져 있다. 상수 k는 0.693/t1/2이고 경과시간 nτ가 만약 t1/2의 4배 또는 5배라면, 지수 -knτ는 -2.8 ~ -3.5로서 e-knτ는 0.03~0.06 정도의 작은 값이 되어 항정상태에 유사하다고 간주한다. 즉, 다회투여 시 4~5회의 반감기가 경과하면 항정상태에 도달하였다고 간주한다. 이 때 흔히, 투여간격 τ를 줄여 자주 투여하면 항정상태에 일찍 도달할 수 있을 것이라는 착각을 할 수 있다. 그러나 앞서 소개된 바와 같이 경과시간 nτ가 t1/2의 4~5배에 도달해야만 항정상태가 되므로 τ를 줄이면 그만큼 n을 늘려줘야만 nτ = t1/2 × 4~5를 달성할 수 있다. 따라서 투여간격과 무관하게 항정상태에 도달하려면 t1/2의 4~5배만큼 시간이 경과해야만 한다. 다만 항정상태 도달시간을 줄이지는 못하지만 τ가 줄어든 만큼 식 (1.3)의 분모 1-e-kτ 값이 작아지므로 얻어지는 Cmax_ss는 커지게 된다. 마찬가지로 투여간격 τ를 늘여도 항정상태 도달시간은 변치 않으며, 1-e-kτ 값이 커지므로 얻어지는 Cmax_ss가 작아지게 된다. 또 t1/2가 τ에 비해 훨씬 짧을 경우 축적 자체가 일어나지 않게 되므로 항정상태라는 개념도 의미가 없게 되는데 수식으로 표현하면 t1/2 × 4~5 < τ 와 같은 상황이 되어 두번째 용량을 투여하기 이전에 이미 항정상태에 도달해 버렸다고도 생각할 수 있다.
1.6 정맥주입(continuous infusion)의 약동학
혈관을 통해 투여하되 한꺼번에 주사(bolus)하지 않고 지속적으로 주입하는 경우, 혈장약물농도는 부드러운 곡선 모양으로 상승하게된다. 이는, 그림 1.13과 같은 다회투여의 약동학을, 용량을 작게 나누어서 매우 짧은 시간간격으로 bolus 주사하는 상황으로 바꾸어 그린 것과 유사하다. 즉, 투여간겨과 용량을 극한까지 작게 만들면 톱니모양 대신 결국 지속주입의 부드러운 농도 곡선을 얻는다. 그러므로 Cmax와 Cmin의 구분없이 부드러운 곡선으로 나온다는 것 외에는 농도가 올라가는 양상이나 항정상태에 도달하는 시간, 주입을 중단하면 1차식을 따라 감소하는 양상 등이 모두 동일하다.
1.7 최종반감기의 개념과 주의점
앞에서는 간결한 설명을 위해 정맥주사 후 말초구획으로 약이 확산되어 급격히 농도가 떨어지는 분포기(그림 1.5)를 가정하지 않았다. 그러나 실제 정맥주사 후 혈장약물농도는 조밀하게 채혈하여 농도를 측정할 경우, 대부분 그림 1.5에 예시된 것처럼 두 개의 기울기가 중첩된 모양을 나타낸다. 분포기의 로그혈장농도 기울기는 훨씬 급격하지만, 대개 짧은 시간 동안만 관찰되므로 이후 이어지는 제거기의 완만한 기울기로써 혈장약물농도의 t1/2를 측정하게 된다. 분포기의 기울기로도 t1/2를 측정할 수 있지만, 구분을 위해 제거기에 얻은 t1/2를 제거반감기, 또는 최종반감기라고 부르며 일반적으로 반감기라는 용어는 최종반감기를 의미한다. 이 최종반감기의 4배가 경과해야 몸에서 약이 대부분 제거된다는 것이 앞에서 언급된 약동학의 기본적 상식이며 대부분의 경우 옳다.
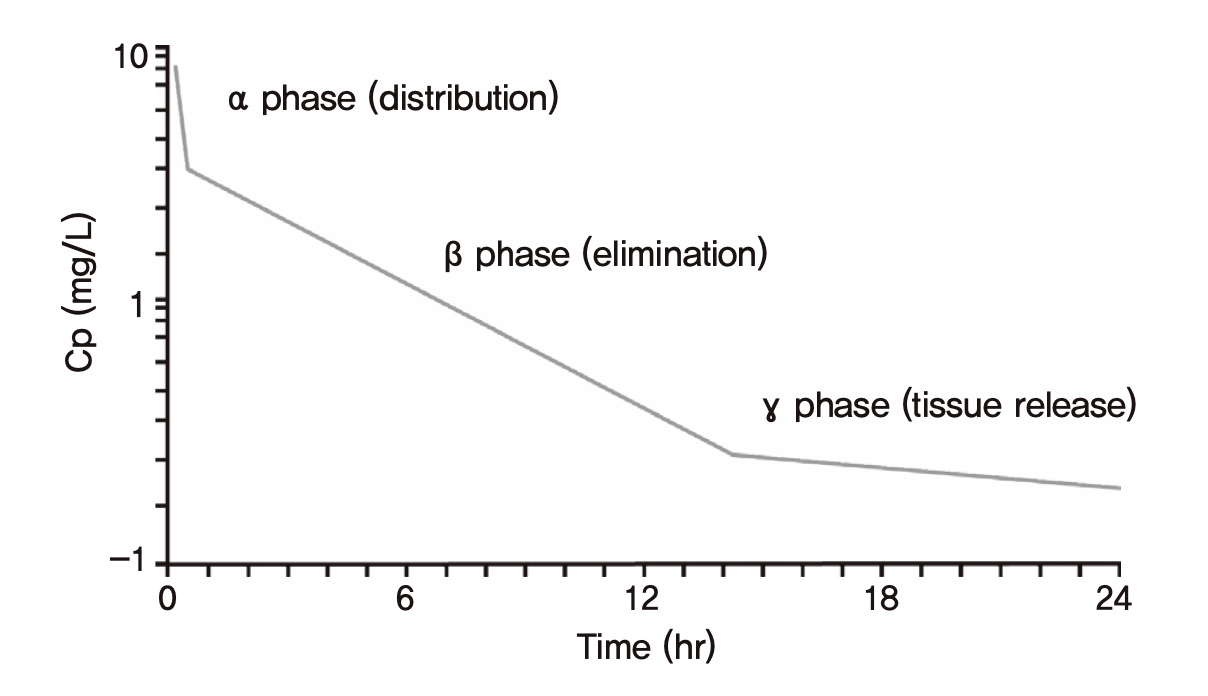
그림 1.14: 3개의 반감기가 관찰되는 경우. 최종반감기는 매우 길지만 몸에 들어온 대부분의 약은 최종 반감기를 보이는 γ-phase 이전에 이미 제거되었으므로 최종반감기의 4배가 지나야 몸에 있는 약물이 대부분 제거된다는 단순한 예상이 어긋나는 사례이다.
그러나 그림 1.14와 같이 혈장농도 곡선에서 여러 개의 반감기를 찾아낼 수 있는 경우, 가장 긴 최종반감기는 별 의미없는 경우가 많다. 예컨데 정맥주사로 투여하는 항암제에서 일주일 이상의 기간동안 PK 채혈을 할 경우 흔히 이런 현상이 관찰된다. 농도가 매우 낮게 떨어진 후 일주일 이상의 최종반감기를 나타내며 길게 꼬리를 끄는 양상을 보이는 경우 약물이 몸에서 제거되는데 4주 이상(최종반감기의 4회 이상)이 걸린다고 판단하면 곤란하다. 몸에 들어온 약은 가장 완만한 기울기를 나타내는 마지막 시기(γ-phase)보다 이전(β-phase)에 이미 대부분 제거되었기 때문이다. 약동학 연구 결과를 분석하는 기본적인 방법인 비구획분석(non-compartmental analysis, NCA)의 결과표에는 이런 자세한 맥락을 담을 수 없고, 그림 1.14와 같은 상황이라면 γ-phase의 반감기가 최종반감기로 기록되는데 그 표에 적힌 반감기 값만 읽으면 이렇게 오판하게 되는 경우가 드물지 않다. 따라서 약동학연구의 결과를 제대로 해석하기 위해서는, 특정한 파라미터 값만 보고 판단하지 말고 그 약물의 약동학적 프로파일을 종합적으로 파악할 수 있어야 한다.
1.8 경구투여의 약동학
지금까지 쉬운 이해를 위해 정맥 bolus injection 상황을 예시하여 설명하였지만, 우리가 아는 약의 99% 이상은 먹는 약이다. 먹는 약이 전신순환혈로 흡수되는데 관련된 약동학 파라미터는 이 장의 처음에 나온대로 ka와 F이다. F가 1이라고 가정하고 1차식에 의해 경구흡수되고 제거되는 약동학을 표현하면 아래 식 (1.4)과 같다.
\[\begin{equation} \frac{{dA}}{{dt}} = \left( - \frac{{dA}_{a}}{{dt}} \right) - kA = k_{a}A_{a} - kA = k_{a}A_{a} - kCV_{d} \tag{1.4} \end{equation}\]
위 식에서 A는 몸 안에 존재하는 약의 양이고 Aa는 소화관 내강(흡수부위)에 남아있는 약의 양으로서 몸에 아직 흡수된 것이 아니므로 몸 안에 있는 약이 아니다. 약을 먹은 직후의 Aa는 물론 Dose와 같으나 시간이 갈수록 줄어들게 된다. \(- \frac{{dA}_{a}}{{dt}}\)는 소화관에 남아있는 약이 줄어드는 속도로서 1차식을 따라 소화관 속에서 몸 안으로 흡수되어 사라짐을 의미한다. 몸 안으로 약이 들어오는 속도는 \(k_{a}A_{a}\)로서 \(\frac{{dA}_{a}}{{dt}}\)와 같은 값이다. 소화관에서 없어지는 속도를 나타날때는 감소하므로 – 부호가 붙어있고 흡수되어 몸 안으로 들어오는 속도를 나타낼때는 +값이다. 몸 안에 있는 약물의 양이 변하는 속도는 몸 안으로 들어오는 속도(\(k_{a}A_{a}\))와 몸 밖으로 빠져나가는 속도(\({kA}\))의 차에 의해 결정됨을 알 수 있다. 즉 약을 먹고 시간이 얼마 안 지났을 때는 Aa의 값이 커서 \(k_{a}A_{a} > kA\) 이므로 \(\frac{{dA}}{{dt}} > 0\) 이고, 혈장약물농도 곡선은 증가하게 된다. 그러다 흡수되는 속도와 제거되는 속도가 같아지는 순간이 오면 \(\frac{{dA}}{{dt}} = 0\) 이고 Cmax에 도달하고 그 시점 이후에는 역전되어 \(\frac{{dA}}{{dt}} < 0\)이 되어 혈장약물농도 곡선은 감소하게 된다. 그러나 Cmax에 도달한 이후 농도가 떨어지기 시작했다고 해서 약의 흡수가 끝난 것은 아니다. \(k_{a}A_{a}\)의 수식으로 표현되는 흡수는 여전히 일어나고 있지만 Aa의 값이 작아서 \(k_{a}A_{a}\)가 \({kA}\) 보다 작을 뿐이다. 흡수가 종료되었다고 할 수 있으려면 충분한 시간이 흘러 Aa가 무시할 수 있을 정도로 작아졌을(0에 가깝게 된) 때이다.
흡수가 비교적 빠르게 일어나는 약이라면 흡수속도상수(ka)가 제거속도상수(k)보다 클 것이고 Cmax에 빠르게 도달하고 Cmax값도 클 것이다. ka가 작아질수록 초기에 농도가 올라가는 경사가 완만해져서 Cmax에 늦게 도달하고 Cmax값도 작아질 것이다. 특히, 약효를 오래 지속시키기 위해 ka를 아주 작게 만드는 기술을 적용하는 제형(서방형)의 약들은 Cmax에도 늦게 도달할 뿐 아니라 이후로도 오랫동안 흡수가 지속되어 마치 약의 t1/2가 늘어난 것처럼 혈장농도가 천천히 떨어지게 만든다. 그러나 앞서 정의한 t1/2는 0.693/k = 0.693 Vd/CL로서 Vd와 CL의 변함이 없이 ka가 바뀌었다고 해서 t1/2가 바뀌어서는 안된다. 경구흡수가 빨리 일어나는 대부분의 약에서는 Cmax에 도달한 후 얼마 지나지 않아 Aa가 빠르게 고갈되므로 그 이후의 약물농도가 떨어지는 기울기는 \(\frac{{dA}}{{dt}} = - kA\) 와 같이 k에 의해서만 결정된다. 서방형의 경우 Aa가 Cmax에 도달한 후 상당히 긴 시간 동안 무시할 수 없는 큰 값을 유지하므로 \(\frac{{dA}}{{dt}} = k_{a}A_{a} - kA\) 로서 몸 안에 존재하는 약의 양 A 또는 그 농도인 C가 떨어지는 기울기는 훨씬 완만하게 표현된다. 즉 몸이 약을 제거하는 능력(CL)은 변치 않지만 오랫동안 소화관에서 몸 속으로 흡수가 계속 되므로 농도가 떨어지는 기울기가 작아지게 되어 반감기가 길어진 것처럼 보이는 것이다. 이러한 반감기를 0.693/k로 정의되는 반감기와 구분하기 위해 겉보기 반감기(apparent half-life)라고 칭한다. 그림 1.15의 맨 오른쪽 곡선이 서방형의 예로 겉보기 반감기가 길어져 있음을 알 수 있다.
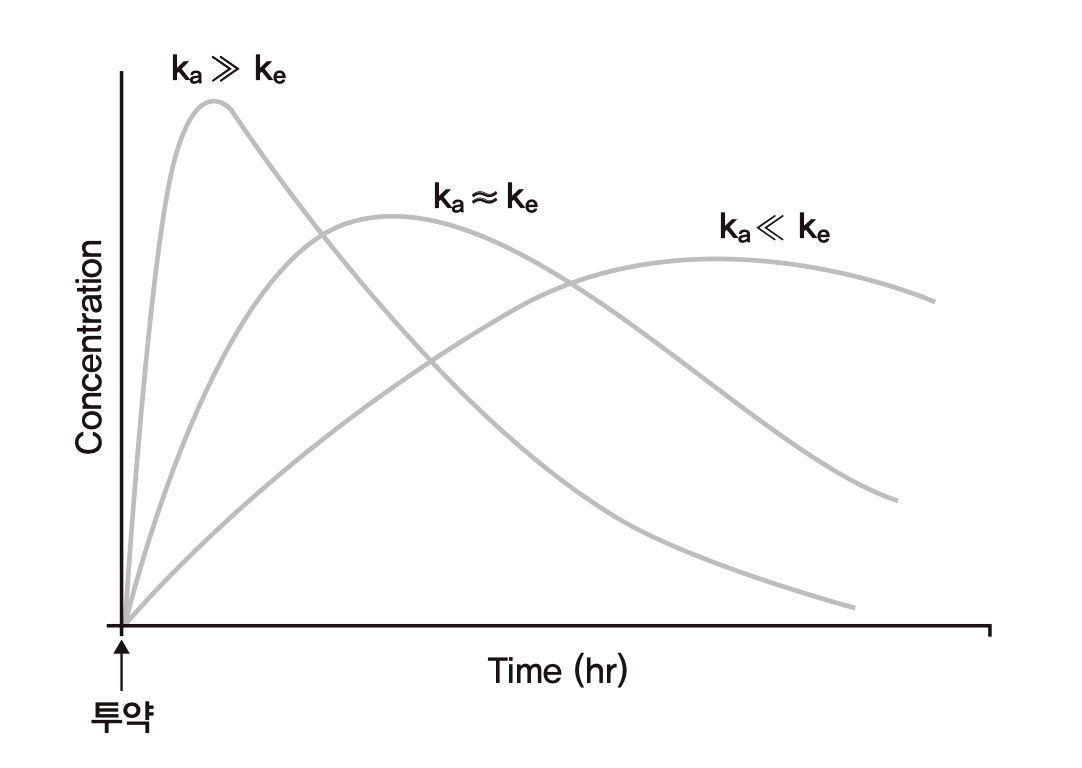
그림 1.15: 흡수속도상수(ka) 와 혈장약물농도 곡선의 관계
1.9 비선형 약동학
1.9.1 비선형약동학과 선형약동학의 차이
이 장에서 지금까지 다룬 약동학은 CL이 일정하고 반감기가 일정한 선형(linear)약동학에 관한 내용이었다. 시판 중인 의약품의 거의 전부가 선형약동학을 따르지만 예외적인 경우가 있는데 phenytoin, salicylate, ethanol 등이 사람 몸에서 비선형(non-linear)약동학을 보이는 물질들이다.
비선형약동학을 이해하기 위해서는 우선 그림 식 (1.5)에 나타난 바와 같이 혈장 약물농도와 대사속도(제거속도)의 관계, CL와의 관계를 살펴보아야 한다. 식 (1.5)의 Vmax와 Km은 약을 대사시키는 장기(간)의 세포 내의 약물대사효소들(다양한 CYP이나 UGT 등)의 특성을 나타내는 값이다. 이에 상응하는 농도는 간 세포 속의 약물농도이지만, 설명의 편의상 혈장약물농도와 동일하다고 가정하였다. 선형약동학을 나타내는 약물은 CL가 혈장약물농도와 상관없이 일정하고, 따라서 약물제거속도는 혈장약물농도가 커질수록 정비례하여 커지게 된다(\(\frac{{dA}}{{dt}} = kA = CL \cdot C(t)\)). 이로 인해 일정한 반감기가 존재한다. 그런데 비선형 약동학의 경우 약물의 제거 속도가 Michelis-Menten(M-M) 수식과 같은 양상을 보여 혈장농도가 높아질수록 약물제거속도가 커지기는 하지만 그 커지는 정도가 점점 둔화되어 포화되는 양상을 보인다.
\[\begin{equation} \frac{{dA}}{{dt}} = \frac{V_{\max} \cdot C}{K_{m} + C} \tag{1.5} \end{equation}\]
간에서 대사되는 약물의 경우 사람에서 얻을 수 있는 약물 농도의 범위가 Km 값보다 무시할만큼 작은 경우에는 선형약동학을 보인다. 즉 식 (1.5)의 분모에서 Km >> C인 경우 \(\frac{{dA}}{{dt}} = \frac{V_{\max} \cdot C}{K_{m} + C} \approx \frac{V_{\max}}{K_{m}}C\) 와 같이 제거속도(\(\frac{{dA}}{{dt}}\))는\(\frac{V_{\max}}{K_{m}}\)에 비례하는데, 결국 CL는 \(\frac{V_{\max}}{K_{m}}\)과 같은 값으로 나타나게 된다. 대부분의 약들의 Km 값은 C 보다 훨씬 커서 선형약동학으로 보이는 것이다. 그런데 농도가 Km에 비해 무시할 수 없게 커지면 약물제거속도는 원래의 수식 (1.5)와 같이 나타나게 되고, 급기야 C가 Km보다 월등히 커지면(Km << C), 식은 \(\frac{{dA}}{{dt}} = \frac{V_{\max} \cdot C}{K_{m} + C} \approx \frac{V_{\max} \cdot C}{C} = V_{\max}\) 와 같이 변형된다. 이 때 수식에서 C가 사라지므로, 0차식(zero-order kinetics)의 양상을 나타낸다고도 표현한다. 이 식은 사람의 몸이 일정 시간 동안에 제거할 수 있는 약의 최대량은 Vmax를 넘어설 수 없음을 나타낸다. 식 (1.5)에서의 농도 C는 사실 세포 내에서의 unbound concentration이라야 하지만 편의상 혈장 약물농도 C의 의미로 사용하였다.
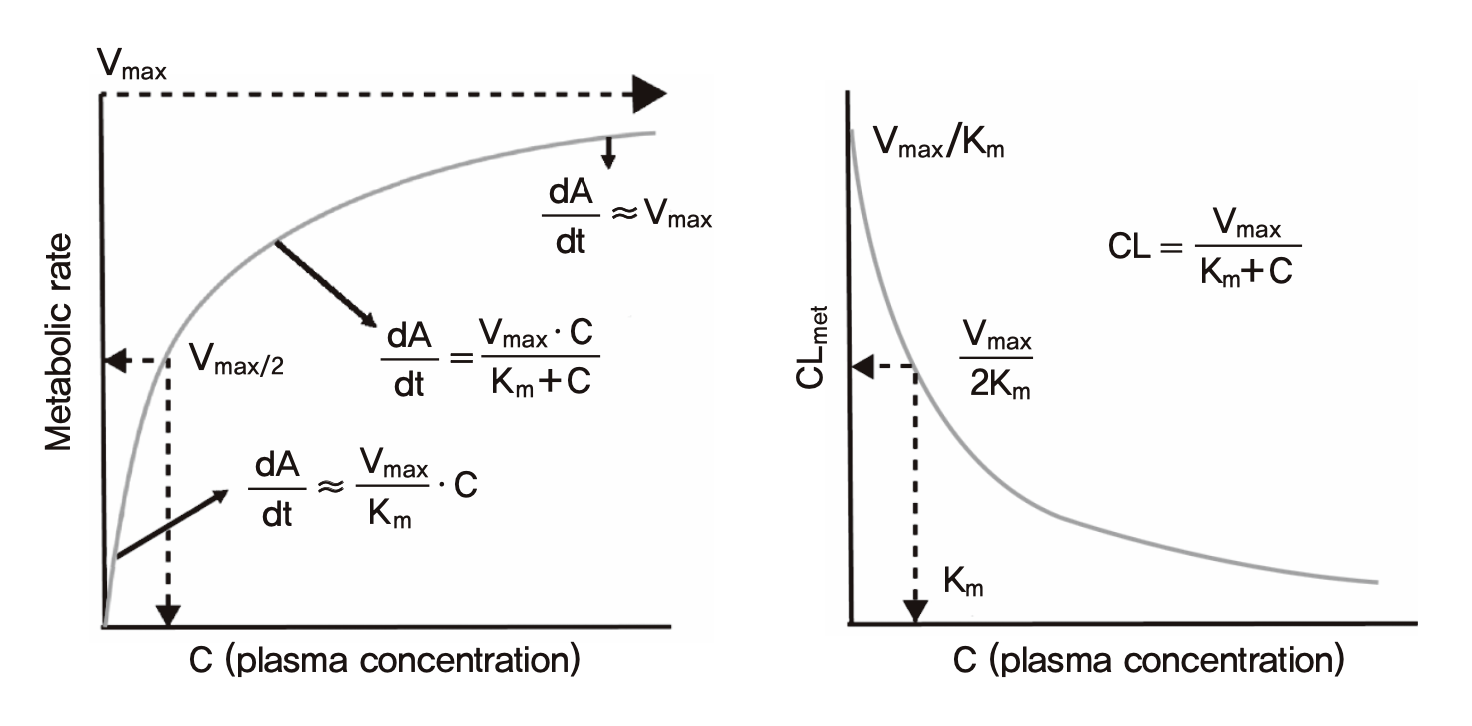
그림 1.16: 비선형약동학을 따르는 약의 혈장농도와 대사속도, CL의 관계
CL = Vmax/(Km+C)이므로 농도가 높아질수록 떨어지고, 반감기 역시 농도가 높아질수록 길어지게 된다. 따라서 매일 투여하는 phenytoin 과 같은 약의 경우, 일일투여용량을 2배로 늘리면 항정상태 혈장농도는 2배보다 훨씬 높게 올라가는 경우가 흔히 발생함이(그림 1.17) 오래전부터 알려져 있다 (Richens and Dunlop 1975). 안전역이 좁은 약들의 경우 비선형약동학을 따르는 경우 용량조절을 위해 혈장농도를 측정하면서 개인별 약동학 파라미터를 추정하는 등 주의를 기울여야 한다. 보통의 선형약동학을 따르는 약물의 경우는 투여량의 증감에 농도도 정비례한다.
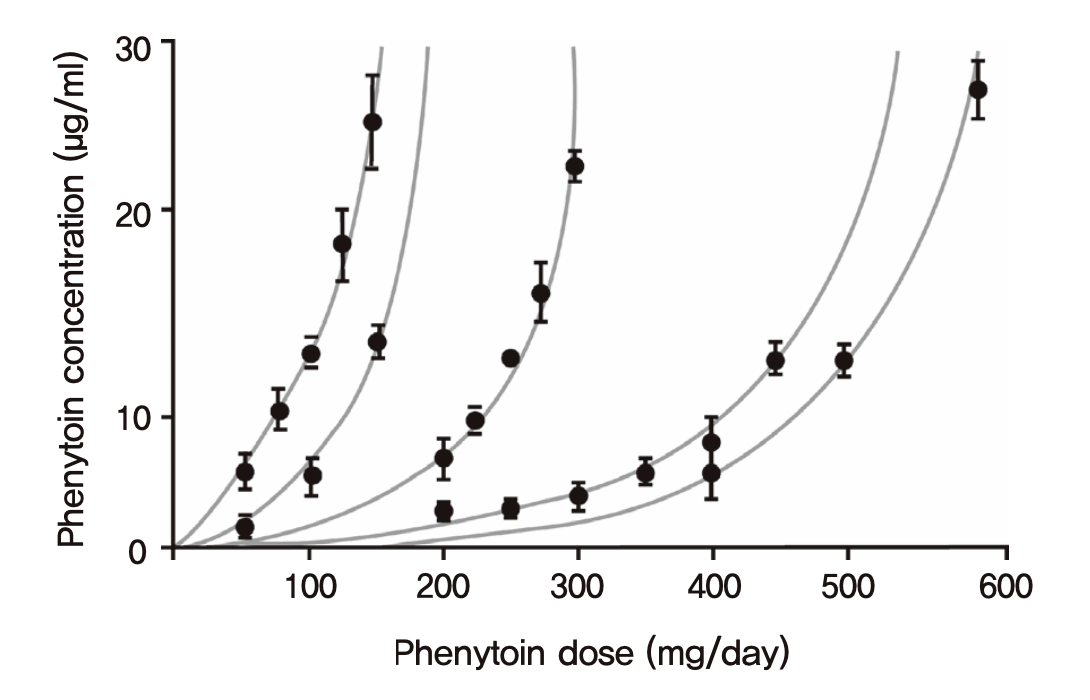
그림 1.17: phenytoin의 혈장농도와 투여량과의 관계. 사람에 따라 기울기는 다르지만, 비선형약동학으로 인해 용량을 늘리는 것보다 농도의 상승이 더욱 가파른 양상을 보인다. (Richens and Dunlop 1975)
혈장약물농도가 시간에 따라 감소하는 경향을 그려보면 로그농도는 볼록한 곡선, 농도는 직선으로 떨어지는 양상을 보이다가 농도가 Km 값보다 훨씬 낮은 수준으로 떨어지면 선형약동학과 같이 로그농도가 직선, 농도가 오목한 곡선모양으로 떨어지게 된다.
1.9.2 비선형약동학과 용량의 관련성
앞서 언급된 바와 같이 선형약동학을 보이는 어떤 약이라 해도, 극단적으로 높은 용량을 투여하면 대사능이 포화상태에 근접하면서 비선형약동학을 나타낼 수 있다. 유사한 상황은 매우 높은 용량까지도 투여하는 비임상 독성시험의 독성약동학(toxicokinetics)자료들에서도 나타날 수 있다.
신약후보물질들의 스크리닝 시 유효농도 부근에서 비선형 약동학을 보이는 물질이라면 가능한 구조변경 등을 통해 선형적인 약동학을 보이는 물질로 대체하는 것이 이후 비선형에 따르는 개발의 숱한 이슈들을 벗어나는 방법이다.
1.10 후보물질의 혈장단백결합율의 해석에 관한 주의할 점
혈장단백질에 결합하지 않은 약물분자들만 타겟에 결합할 수 있다는 것이 이른바 free drug hypothesis이다. 어떤 용량용법으로 사람에게 투여된 약이 효과를 나타내기 위해서는 그 용량용법으로 연속 투여한 후 얻어지는 항정상태에서의 혈장 비결합약물농도(Css,u = fu · Ctot)가 IC50, EC50, Km 등의 약력학 파라미터의 값보다 투여간격 동안 더 높게 유지되어야 한다고 볼 수 있다. 충분히 높은 혈장에서의 비결합농도(free concentration)를 확보한다는 측면에서 후보물질의 물리화학적 특성 중 혈장단백결합율이 낮은, 즉 fu가 높은 물질일수록 유리할 것이라고 흔히 짐작함으로 인해, 후보물질의 초기 스크리닝에 있어 fu가 낮은 물질을 제외시키려는 생각을 하기도 한다. 그러나 실제 시판 중인 약 성분들의 30% 가량이 혈장단백결합율이 95%이상(즉 fu < 0.05)임을 감안하면, 단순히 fu가 낮다고 좋은 후보물질이 아니라는 우려는 지나친 것이다. 중요한 것은 치료용량에서 확보되는 Css,u인데 이는 fu 뿐만 아니라 다른 여러 PK 파라미터들에 의해 결정되기 때문이다. 또한, in vitro에서 구한 IC50, EC50, Km 등은 임상시험에서 효과를 나타내는 혈장약물농도보다 훨씬 높은 경우도 약물 군별로 보고된 바 있다. (즉, in vitro IC50, EC50, Km 보다 훨씬 낮은 Css,u에서도 사람에서 충분한 약효가 나타나는 경우들: (Yim 2019)) 그러므로 비임상 개발 단계에서는 in vitro 약효실험 데이터와 fu 만으로 유효농도를 추정할 수도 있지만, 질병모델동물에서 in vivo 약효실험을 수행한 후 얻어진 약효와 혈장농도의 관계로부터 사람에서의 유효용량이나 유효혈장농도를 추정하는 것이 좀 더 신뢰할 수 있는 접근법이다.
1.11 맺음말
일정한 CL와 t1/2를 나타내는 약물에서의 약동학인 선형약동학은 의약품의 체내 거동을 이해하기 위한 가장 중요하고 기본적인 도구이다. Vd와 CL에 의해 k와 반감기가 결정되고, 흡수는 ka와 F에 의해 결정된다. 이 네 가지 파라미터와 투여량(Dose), 투여 후 경과 시간(t)의 함수로 표현되는 것이 혈장약물농도이다. 동물이나 사람에서 약동학 시험을 거쳐 얻어진 혈장약물농도와 시간, 용량의 값들로부터 AUC와 t1/2를 계산하고 이로부터 Vd와 CL를 구하는 것이 흔히 쓰이는 NCA 방법이다. NCA 방법은 비교적 단순하고, 쉽게 약동학적 프로파일을 알 수 있게 해준다는 장점이 있으나 약동학 자료로부터 얻을 수 있는 보다 많은 정보들을 놓치게 되므로 이를 보완하기 위해 구획분석법(compartmental analysis)이 함께 쓰이고 있다.
비선형 약동학이지만 M-M과도 다른 양상을 보이는 예로는 단클론항체의 약동학이 있다. 단클론항체는 타겟이 되는 항원이 혈액 내에 존재하는지, 세포표면에 존재하는지, 결합 후 항원세포내로 이동하는지 등에 따라 훨씬 다양하고 복잡한 양상을 나타낸다.